

Obesity Associated with Prediabetes Increases the Risk of Breast Cancer Development and Progression—A Study on an Obese Rat Model with Impaired Glucose Tolerance


 ,
,
Abstract
1. Introduction
2. Results
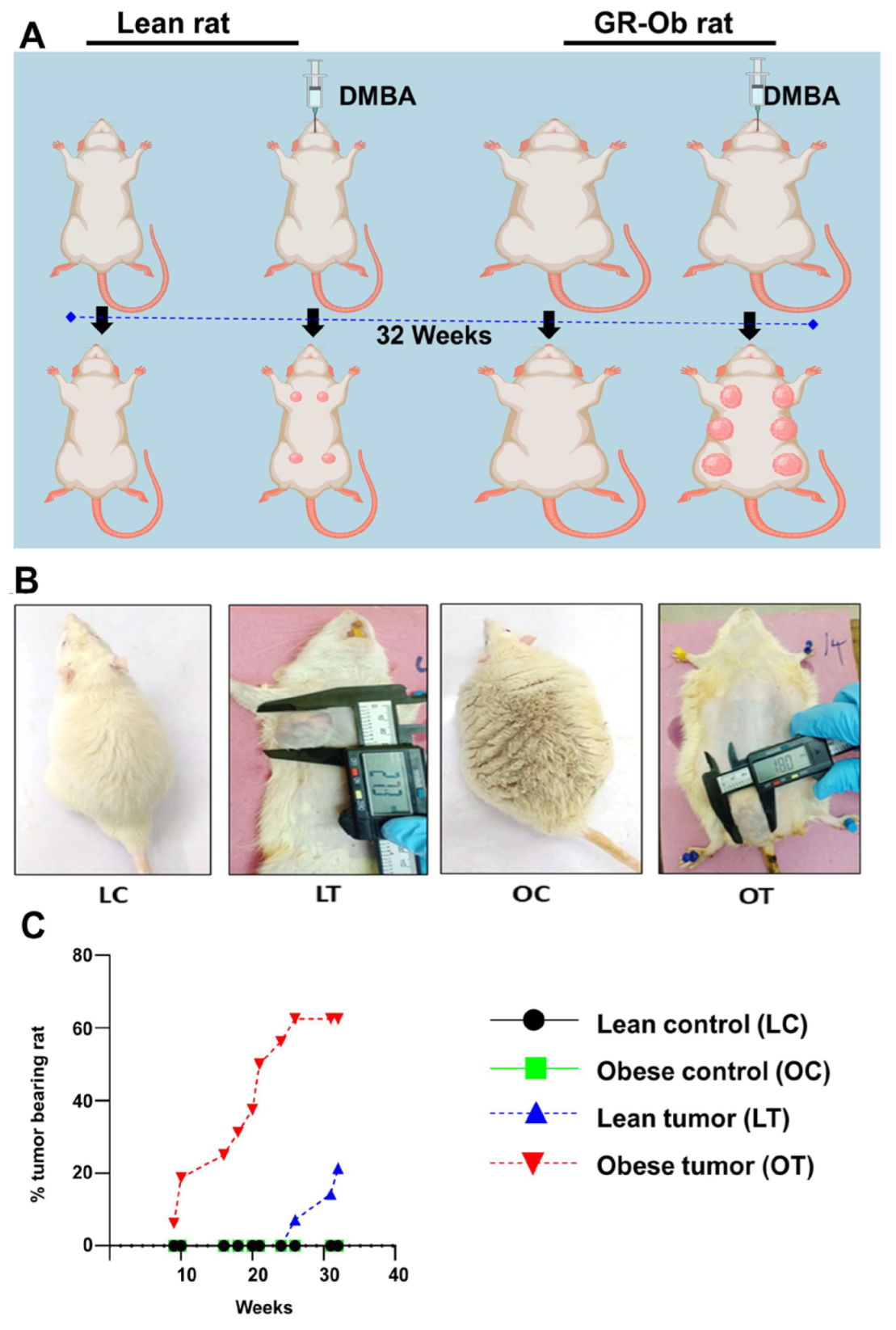
2.1. Obesity Accelerates the Onset and Development of Breast Cancer
2.2. Obesity Promotes Oncogenic Markers Expression in Rat Mammary Gland
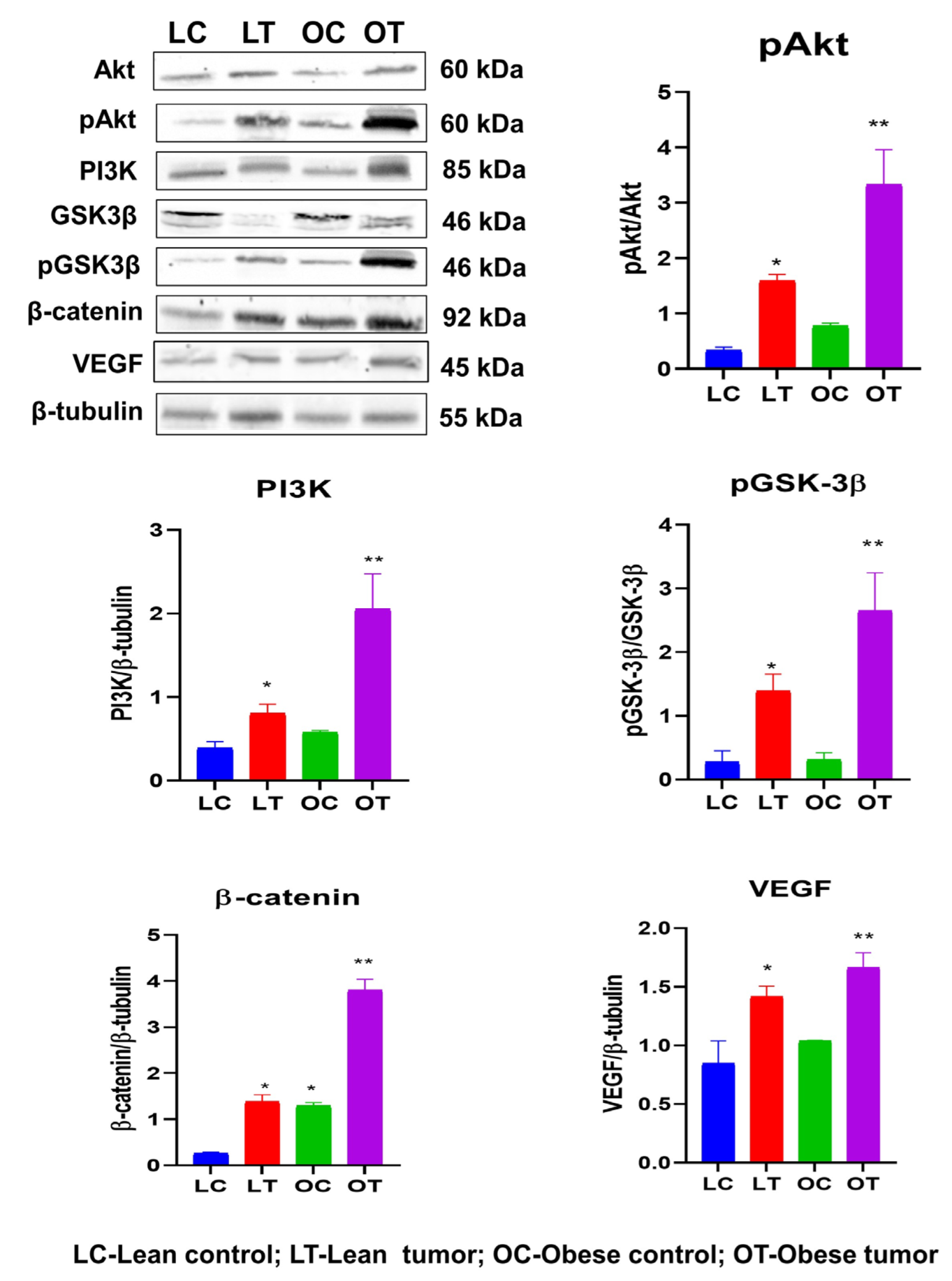
2.3. Immunohistochemistry of Insulin Signaling Pathway and PI3K/GSK3β/β-Catenin Role in Breast Cancer Development
3. Discussion
4. Materials and Methods
4.1. Animal Grouping and Housing
4.2. DMBA Preparation and Administration
4.3. Measurements of Tumor Growth Parameters
4.4. Gross Necropsy
4.5. Hematoxylin and Eosin Staining
4.6. Immunohistochemistry
4.7. Immunoblotting
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Chun, M.; Oh, Y.T.; Noh, O.K.; Kim, L. Metabolic Comorbidities and Medical Institution Utilization among Breast Cancer Survivors: A National Population-based Study. Korean J. Intern. Med. 2020, 35, 421–428. [Google Scholar] [CrossRef]
- Buono, G.; Crispo, A.; Giuliano, M.; De Angelis, C.; Schettini, F.; Forestieri, V.; Lauria, R.; De Laurentiis, M.; De Placido, P.; Rea, C.G.; et al. Metabolic Syndrome and Early-Stage Breast Cancer Outcome: Results from a Prospective Observational Study. Breast Cancer Res. Treat. 2020, 182, 401–409. [Google Scholar] [CrossRef]
- Kennard, K.; Buckley, M.E.; Sizer, L.M.; Larson, S.; Carter, W.B.; Frazier, T.G.; Carp, N.Z. Metabolic Syndrome: Does This Influence Breast Cancer Outcomes in the Triple-Negative Population? Breast Cancer Res. Treat. 2021, 186, 53–63. [Google Scholar] [CrossRef] [PubMed]
- The World Health Organization. Obesity and Overweight Key Facts. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 23 June 2023).
- The World Obesity Federation (World Obesity). Available online: https://www.worldobesity.org/resources/resource-library/world-obesity-atlas-2022 (accessed on 23 June 2023).
- International Diabetes Federation (IDF) Diabetes Atlas. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 23 June 2023).
- Wilson, M.L. Prediabetes: Beyond the Borderline. Nurs. Clin. N. Am. 2017, 52, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Ennis, C.S.; Llevenes, P.; Qiu, Y.; Dries, R.; Denis, G.V. The Crosstalk within the Breast Tumor Microenvironment in Type II Diabetes: Implications for Cancer Disparities. Front. Endocrinol. 2022, 13, 1044670. [Google Scholar] [CrossRef]
- Lu, Y.; Hajjar, A.; Cryns, V.L.; Trentham-Dietz, A.; Gangnon, R.E.; Heckman-Stoddard, B.M.; Alagoz, O. Breast Cancer Risk for Women with Diabetes and the Impact of Metformin: A Meta-analysis. Cancer Med. 2022, 12, 11703–11718. [Google Scholar] [CrossRef]
- Luís, C.; Dias, J.; Firmino-Machado, J.; Fernandes, R.; Pereira, D.; Baylina, P.; Fernandes, R.; Soares, R. A Retrospective Study in Tumour Characteristics and Clinical Outcomes of Overweight and Obese Women with Breast Cancer. Breast Cancer Res. Treat. 2023, 198, 89–101. [Google Scholar] [CrossRef]
- Pati, S.; Irfan, W.; Jameel, A.; Ahmed, S.; Shahid, R.K. Obesity and Cancer: A Current Overview of Epidemiology, Pathogenesis, Outcomes, and Management. Cancers 2023, 15, 485. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.; Bae, S.J.; Ji, J.H.; Lee, J.W.; Son, B.H.; Ahn, S.H.; Jeong, J.; Lee, S.B.; Ahn, S.G. Association of Body Mass Index with 21-Gene Recurrence Score among Women with Estrogen Receptor-Positive, ERBB2-Negative Breast Cancer. JAMA Netw. Open 2022, 5, e2243935. [Google Scholar] [CrossRef] [PubMed]
- Calip, G.S.; Yu, O.; Boudreau, D.M.; Shao, H.; Oratz, R.; Richardson, S.B.; Gold, H.T. Diabetes and Differences in Detection of Incident Invasive Breast Cancer. Cancer Causes Control 2019, 30, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Arce, L.; Robles-Rodríguez, N.; Fernández-Feito, A.; Llaneza-Folgueras, A.; Encinas-Muñiz, A.I.; Lana, A. Type 2 Diabetes and all-cause mortality among Spanish women with breast cancer. Cancer Causes Control 2022, 33, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Gill, A.A.; Zahm, S.H.; Jatoi, I.; Shriver, C.D.; McGlynn, K.A.; Zhu, K. Diabetes and Overall Survival among Breast Cancer Patients in the U.S. Military Health System. Cancer Epidemiol. Biomark. Prev. 2018, 27, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Jacob, L.; Kostev, K.; Rathmann, W.; Kalder, M. Impact of Metformin on Metastases in Patients with Breast Cancer and Type 2 Diabetes. J. Diabetes Its Complicat. 2016, 30, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Crispo, A.; Augustin, L.S.; Grimaldi, M.; Nocerino, F.; Giudice, A.; Cavalcanti, E.; Di Bonito, M.; Botti, G.; De Laurentiis, M.; Rinaldo, M.; et al. Risk Differences Between Prediabetes and Diabetes According to Breast Cancer Molecular Subtypes. J. Cell. Physiol. 2017, 232, 1144–1150. [Google Scholar] [CrossRef]
- Bernard, J.J.; Wellberg, E.A. The Tumor Promotional Role of Adipocytes in the Breast Cancer Microenvironment and Macroenvironment. Am. J. Pathol. 2021, 191, 1342–1352. [Google Scholar] [CrossRef]
- Maskarinec, G.; Jacobs, S.; Park, S.Y.; Haiman, C.A.; Setiawan, V.W.; Wilkens, L.R.; Le Marchand, L. Type II Diabetes, Obesity, and Breast Cancer Risk: The Multiethnic Cohort. Cancer Epidemiol. Biomark. Prev. 2017, 26, 854–861. [Google Scholar] [CrossRef]
- Maskarinec, G.; Sadakane, A.; Sugiyama, H.; Brenner, A.; Tatsukawa, Y.; Grant, E. Type 2 Diabetes, Obesity, and Breast Cancer Risk among Japanese Women of the Atomic Bomb Survivor Cohort. Cancer Epidemiol. 2019, 60, 179–184. [Google Scholar] [CrossRef]
- Maskarinec, G.; Haraldsdóttir, Á.; Einarsdóttir, K.; Aspelund, T.; Tryggvadóttir, L.; Harris, T.B.; Gudnason, V.; Torfadóttir, J.E. Type 2 Diabetes and Obesity in Midlife and Breast Cancer Risk in the Reykjavik Cohort. Cancer Causes Control 2019, 30, 1057–1065. [Google Scholar] [CrossRef]
- McTiernan, A. Diet and Prognosis in Women with Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2021, 30, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, X.; Qiu, M.; Chen, P.; Tang, H.; Hu, Y.; Huang, Y. Prediabetes and the risk of cancer: A meta-analysis. Diabetologia 2014, 57, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Luque, R.M.; López-Sánchez, L.M.; Villa-Osaba, A.; Luque, I.M.; Santos-Romero, A.L.; Yubero-Serrano, E.M.; Cara-García, M.; Álvarez-Benito, M.; López-Mirand, A.J.; Gahete, M.D.; et al. Breast Cancer is Associated to Impaired Glucose/Insulin Homeostasis in Premenopausal Obese/Overweight Patients. Oncotarget 2017, 8, 81462–81474. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.H.; LeRoith, D. Obesity, Type 2 Diabetes, and Cancer: The Insulin and IGF Connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef] [PubMed]
- Harishankar, N.; Vajreswari, A.; Giridharan, N.V. WNIN/GR-Ob—An Insulin-Resistant Obese Rat Model from Inbred WNIN Strain. Indian J. Med. Res. 2011, 134, 320–329. [Google Scholar] [PubMed]
- Godisela, K.K.; Reddy, S.S.; Kumar, C.U.; Saravanan, N.; Reddy, P.Y.; Jablonski, M.M.; Ayyagari, R.; Reddy, G.B. Impact of Obesity with Impaired Glucose Tolerance on Retinal Degeneration in a Rat Model of Metabolic Syndrome. Mol. Vis. 2017, 23, 263–274. [Google Scholar]
- Schlesinger, S.; Neuenschwander, M.; Barbaresko, J.; Lang, A.; Maalmi, H.; Rathmann, W.; Roden, M.; Herder, C. Prediabetes and Risk of Mortality, Diabetes-Related Complications and Comorbidities: Umbrella Review of Meta-Analyses of Prospective Studies. Diabetologia 2022, 65, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.M.; Jaradat, S.K.; Alhusban, A.; Tahaineh, L. Glycosylated Hemoglobin A1c Is Associated with Anthropometric Measurements and Tumor Characteristics in Breast Cancer Patients. Int. J. Women’s Health 2020, 12, 139–149. [Google Scholar] [CrossRef]
- Cao, Y.; Xia, B.; Zhang, Z.; Hu, D.; Huang, X.; Yuan, J.; Li, F. Association of Body Fat Distribution and Risk of Breast Cancer in Pre- and Postmenopausal Women. Obes. Facts 2023, 1–8. [Google Scholar] [CrossRef]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef]
- Belardi, V.; Gallagher, E.J.; Novosyadlyy, R.; LeRoith, D. Insulin and IGFs in Obesity-Related Breast Cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Ecker, B.L.; Lee, J.Y.; Sterner, C.J.; Solomon, A.C.; Pant, D.K.; Shen, F.; Peraza, J.; Vaught, L.; Mahendra, S.; Belka, G.K.; et al. Impact of Obesity on Breast Cancer Recurrence and Minimal Residual Disease. Breast Cancer Res. 2019, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Cahyono, R.; Prabowo, D.; Avanti, W.S.; Choridah, L.; Dwianingsih, E.K.; Harahap, W.A.; Aryandono, T. Metabolic Comorbidities and the Association with Risks of Recurrent Metastatic Disease in Breast Cancer Survivors. BMC Cancer 2021, 21, 590. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Agullo, P.; Lakshmanaswamy, R. Links between Obesity, Diabetes and Ethnic Disparities in Breast Cancer among Hispanic Populations. Obes. Rev. 2013, 14, 679–691. [Google Scholar] [CrossRef]
- Monzavi-Karbassi, B.; Gentry, R.; Kaur, V.; Siegel, E.R.; Jousheghany, F.; Medarametla, S.; Fuhrman, B.J.; Safar, A.M.; Hutchins, L.F.; Kieber-Emmons, T. Pre-Diagnosis Blood Glucose and Prognosis in Women with Breast Cancer. Cancer Metab. 2016, 4, 7. [Google Scholar] [CrossRef]
- Vella, V.; Lappano, R.; Bonavita, E.; Maggiolini, M.; Clarke, R.B.; Belfiore, A.; De Francesco, E.M. Insulin/IGF Axis and the Receptor for Advanced Glycation End Products: Role in Meta-Inflammation and Potential in Cancer Therapy. Endocr. Rev. 2023. [Google Scholar] [CrossRef]
- Wolff, G.L.; Kodell, R.L.; Cameron, A.M.; Medina, D. Accelerated Appearance of Chemically Induced Mammary Carcinomas in Obese Yellow (Avy/A) (BALB/c X VY) F1 Hybrid Mice. J. Toxicol. Environ. Health 1982, 10, 131–142. [Google Scholar] [CrossRef]
- Hakkak, R.; Holley, A.W.; Macleod, S.L.; Simpson, P.M.; Fuchs, G.J.; Jo, C.H.; Kieber-Emmons, T.; Korourian, S. Obesity Promotes 7,12-dimethylbenz(a)anthracene-induced Mammary Tumor Development in Female Zucker Rats. Breast Cancer Res. 2005, 7, R627. [Google Scholar] [CrossRef]
- Hakkak, R.; MacLeod, S.; Shaaf, S.; Holley, A.W.; Simpson, P.; Fuchs, G.; Jo, C.H.; Kieber-Emmons, T.; Korourian, S. Obesity Increases the Incidence of 7,12-dimethylbenz(a)anthracene-Induced Mammary Tumors in an Ovariectomized Zucker Rat model. Int. J. Oncol. 2007, 30, 557–563. [Google Scholar] [CrossRef]
- Kabel, A.M. Tumor Markers of Breast Cancer: New prospectives. J. Oncol. Sci. 2017, 3, 5–11. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Au, C.C.; Benito-Martin, A.; Ladumor, H.; Oshchepkova, S.; Moges, R.; Brown, K.A. Estrogens and Breast Cancer: Mechanisms Involved in Obesity-Related Development, Growth and Progression. J. Steroid Biochem. Mol. Biol. 2019, 189, 161–170. [Google Scholar] [CrossRef]
- Chauhan, R.; Trivedi, V.; Rani, R.; Singh, U. A Comparative Analysis of Body Mass Index with Estrogen Receptor, Progesterone Receptor and Human Epidermal Growth Factor Receptor 2 Status in Pre- and Postmenopausal Breast Cancer Patients. J. Midlife Health 2020, 11, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Oudanonh, T.; Nabi, H.; Ennour-Idrissi, K.; Lemieux, J.; Diorio, C. Progesterone Receptor Status Modifies the Association Between Body Mass Index and Prognosis in Women Diagnosed with Estrogen Receptor Positive Breast Cancer. Int. J. Cancer 2020, 146, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- de Azambuja, E.; Cardoso, F.; de Castro, G.; Colozza, M.; Mano, M.S.; Durbecq, V.; Sotiriou, C.; Larsimont, D.; Piccart-Gebhart, M.J.; Paesmans, M. Ki-67 as Prognostic Marker in Early Breast Cancer: A Meta-analysis of Published Studies Involving 12,155 Patients. Br. J. Cancer 2007, 96, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Inic, Z.; Zegarac, M.; Inic, M.; Markovic, I.; Kozomara, Z.; Djurisic, I.; Inic, I.; Pupic, G.; Jancic, S. Difference between Luminal A and Luminal B Subtypes According to Ki-67, Tumor Size, and Progesterone Receptor Negativity Providing Prognostic Information. Clinical Medicine Insights. Oncology 2014, 8, 107–111. [Google Scholar] [CrossRef]
- Skjervold, A.H.; Pettersen, H.S.; Valla, M.; Opdahl, S.; Bofin, A.M. Visual and Digital Assessment of Ki-67 in Breast Cancer Tissue—A Comparison of Methods. Diagn. Pathol. 2022, 17, 45. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and Degradation of Mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef]
- Yang, P.; Du, C.W.; Kwan, M.; Liang, S.X.; Zhang, G.J. The Impact of p53 in Predicting Clinical Outcome of Breast Cancer Patients with Visceral Metastasis. Sci. Rep. 2013, 3, 2246. [Google Scholar] [CrossRef]
- Alsner, J.; Yilmaz, M.; Guldberg, P.; Hansen, L.L.; Overgaard, J. Heterogeneity in the Clinical Phenotype of TP53 Mutations in Breast Cancer Patients. Breast Cancer Res. 2000, 2, P4.04. [Google Scholar] [CrossRef]
- Jasar, D.; Smichkoska, S.; Kubelka, K.; Filipovski, V.; Petrushevska, G. Expression of p53 Protein Product in Triple Negative Breast Cancers and Relation with Clinical and Histopathological Parameters. Prilozi 2015, 36, 69–79. [Google Scholar] [CrossRef]
- Bae, S.Y.; Nam, S.J.; Jung, Y.; Lee, S.B.; Park, B.-W.; Lim, W.; Jung, S.H.; Yang, H.W.; Jung, S.P. Differences in Prognosis and Efficacy of Chemotherapy by p53 Expression in Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2018, 172, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, X.; Wang, Y.; Sun, G.; Zhang, J.; Li, Z. Obesity and Endocrine-Related Cancer: The Important Role of IGF-1. Front. Endocrinol. 2023, 14, 1093257. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; Johnson, T.; Tikk, K.; Sookthai, D.; Tjønneland, A.; Roswall, N.; Overvad, K.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; Dossus, L.; et al. Insulin-Like Growth Factor I and Risk of Breast Cancer by Age and Hormone Receptor Status—A Prospective Study within the EPIC Cohort. Int. J. Cancer 2014, 134, 2683–2690. [Google Scholar] [CrossRef]
- Murphy, N.; Knuppel, A.; Papadimitriou, N.; Martin, R.M.; Tsilidis, K.K.; Smith-Byrne, K.; Fensom, G.; Perez-Cornago, A.; Travis, R.C.; Key, T.J.; et al. Insulin-Like Growth Factor-1, Insulin-Like Growth Factor-Binding Protein-3, and Breast Cancer Risk: Observational and Mendelian Randomization Analyses with ~430 000 Women. Ann. Oncol. 2020, 31, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front. Endocrinol. 2015, 6, 59. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The Role of the Insulin-Like Growth Factor-1 System in Breast Cancer. Mol. Cancer 2015, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Monson, K.R.; Goldberg, M.; Wu, H.-C.; Santella, R.M.; Chung, W.K.; Terry, M.B. Circulating Growth Factor Concentrations and Breast Cancer Risk: A Nested Case-Control Study of IGF-1, IGFBP-3, and Breast Cancer in a Family-Based Cohort. Breast Cancer Res. 2020, 22, 109. [Google Scholar] [CrossRef]
- Kahlert, S.; Nuedling, S.; van Eickels, M.; Vetter, H.; Meyer, R.; Grohe, C. Estrogen Receptor Alpha Rapidly Activates the IGF-1 Receptor Pathway. J. Biol. Chem. 2000, 275, 18447–18453. [Google Scholar] [CrossRef]
- Yoshimaru, T.; Komatsu, M.; Miyoshi, Y.; Honda, J.; Sasa, M.; Katagiri, T. Therapeutic Advances in BIG3-PHB2 Inhibition Targeting the Crosstalk between Estrogen and Growth Factors in Breast Cancer. Cancer Sci. 2015, 106, 550–558. [Google Scholar] [CrossRef]
- Fox, E.M.; Kuba, M.G.; Miller, T.W.; Davies, B.R.; Arteaga, C.L. Autocrine IGF-I/insulin Receptor Axis Compensates for Inhibition of AKT in ER-Positive Breast Cancer Cells with Resistance to Estrogen Deprivation. Breast Cancer Res. 2013, 15, R55. [Google Scholar] [CrossRef]
- Giles, E.D.; Wellberg, E.A. Preclinical Models to Study Obesity and Breast Cancer in Females: Considerations, Caveats, and Tools. J. Mammary Gland Biol. Neoplasia 2020, 25, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, P.; Zhang, A.; Mao, X. Advances in Rodent Models for Breast Cancer Formation, Progression, and Therapeutic Testing. Front. Oncol. 2021, 11, 593337. [Google Scholar] [CrossRef] [PubMed]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a Breast Cancer Target. npj Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K Signaling in Cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Sefidbakht, S.; Saeedipour, H.; Saffar, H.; Mirzaian, E. Determination of β-catenin Expression in Breast Cancer and Its Relationship with Clinicopathologic Parameters. Asian Pac. J. Cancer Prev. 2021, 22, 3493–3498. [Google Scholar]
- Yuan, Y.; Long, H.; Zhou, Z.; Fu, Y.; Jiang, B. PI3K-AKT-Targeting Breast Cancer Treatments: Natural Products and Synthetic Compounds. Biomolecules 2023, 13, 93. [Google Scholar] [CrossRef]
- Li, S.; Li, S.; Sun, Y.; Li, L. The Expression of β-catenin in Different Subtypes of Breast Cancer and its Clinical Significance. Tumor Biol. 2014, 35, 7693–7698. [Google Scholar] [CrossRef]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is Required for the Tumorigenic Behavior of Triple-Negative Breast Cancer Cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt Signaling in Breast Cancer: Biological Mechanisms, Challenges and Opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Bojková, B.; Kajo, K.; Kisková, T.; Kubatka, P.; Žúbor, P.; Solár, P.; Péč, M.; Adamkov, M. Metformin and Melatonin Inhibit DMBA-Induced Mammary Tumorigenesis in Rats Fed a High-Fat Diet. Anticancer Drugs 2018, 29, 128–135. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Rasband, W.S. ImageJ. National Institutes of Health, Bethesda, Maryland, USA. 1997–2015. Available online: https://imagej.nih.gov/ij/ (accessed on 19 June 2023).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | DMBA-Treated | |||
|---|---|---|---|---|
| Lean | Obese | Lean | Obese | |
| Incidence percentage | 0 | 0 | 21.42 | 62.5 |
| Latency period | 0 | 0 | 211 | 119 |
| Average tumor volume (cm2) | 0 | 0 | 5.5 | 3.06 |
| Cumulative tumor volume (cm2) | 0 | 0 | 22.22 | 42.84 |
| Total Number of tumors | 0 | 0 | 4 | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kallamadi, P.R.; Esari, D.; Addi, U.R.; Kesavan, R.; Putcha, U.K.; Nagini, S.; Reddy, G.B. Obesity Associated with Prediabetes Increases the Risk of Breast Cancer Development and Progression—A Study on an Obese Rat Model with Impaired Glucose Tolerance. Int. J. Mol. Sci. 2023, 24, 11441. https://doi.org/10.3390/ijms241411441
Kallamadi PR, Esari D, Addi UR, Kesavan R, Putcha UK, Nagini S, Reddy GB. Obesity Associated with Prediabetes Increases the Risk of Breast Cancer Development and Progression—A Study on an Obese Rat Model with Impaired Glucose Tolerance. International Journal of Molecular Sciences. 2023; 24(14):11441. https://doi.org/10.3390/ijms241411441
Chicago/Turabian StyleKallamadi, Prathap Reddy, Deepshika Esari, Utkarsh Reddy Addi, Rushendhiran Kesavan, Uday Kumar Putcha, Siddavaram Nagini, and Geereddy Bhanuprakash Reddy. 2023. "Obesity Associated with Prediabetes Increases the Risk of Breast Cancer Development and Progression—A Study on an Obese Rat Model with Impaired Glucose Tolerance" International Journal of Molecular Sciences 24, no. 14: 11441. https://doi.org/10.3390/ijms241411441
APA StyleKallamadi, P. R., Esari, D., Addi, U. R., Kesavan, R., Putcha, U. K., Nagini, S., & Reddy, G. B. (2023). Obesity Associated with Prediabetes Increases the Risk of Breast Cancer Development and Progression—A Study on an Obese Rat Model with Impaired Glucose Tolerance. International Journal of Molecular Sciences, 24(14), 11441. https://doi.org/10.3390/ijms241411441