
Artificial Intelligence in Decrypting Cytoprotective Activity under Oxidative Stress from Molecular Structure
 , ,
, ,  ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
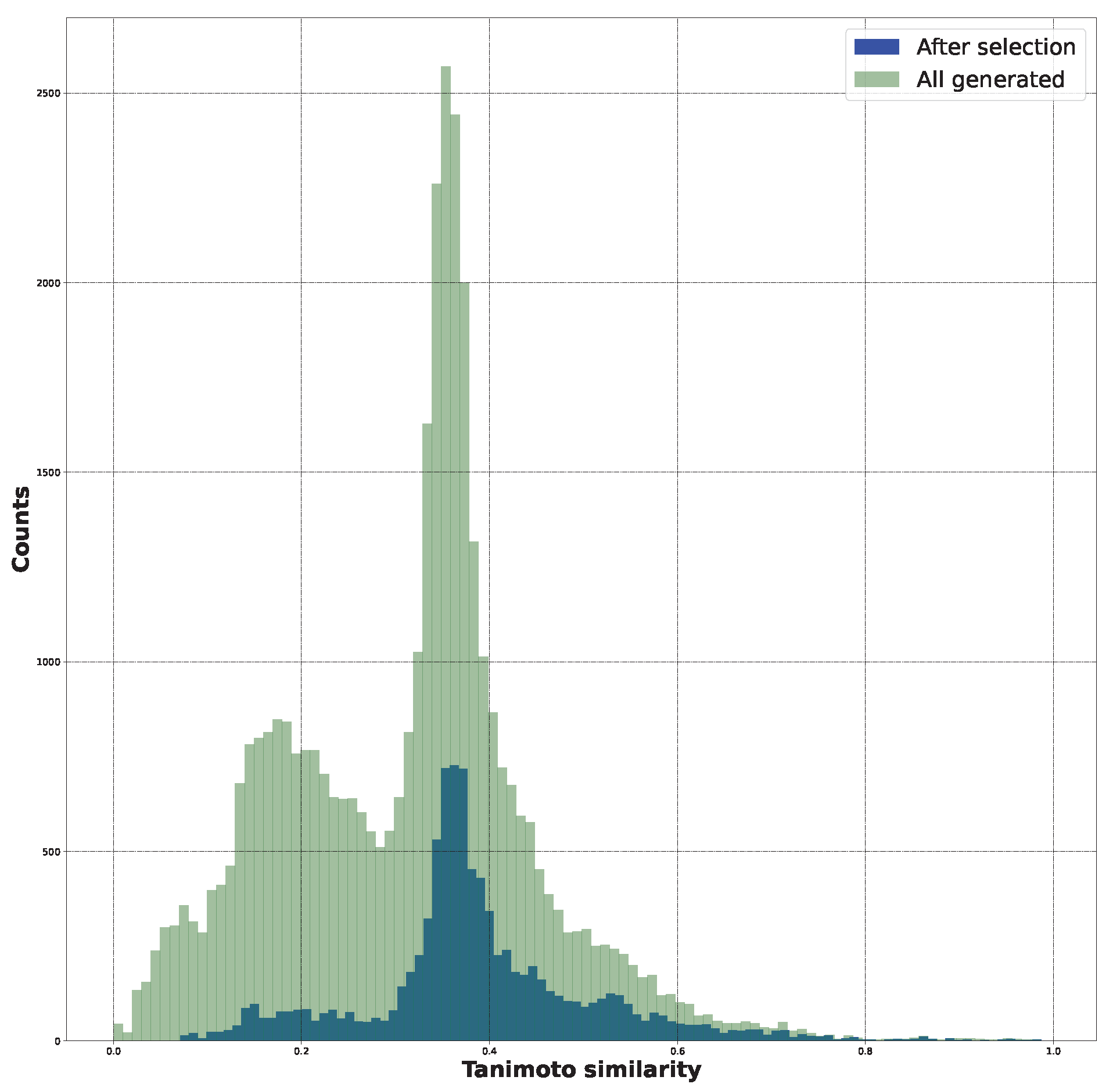
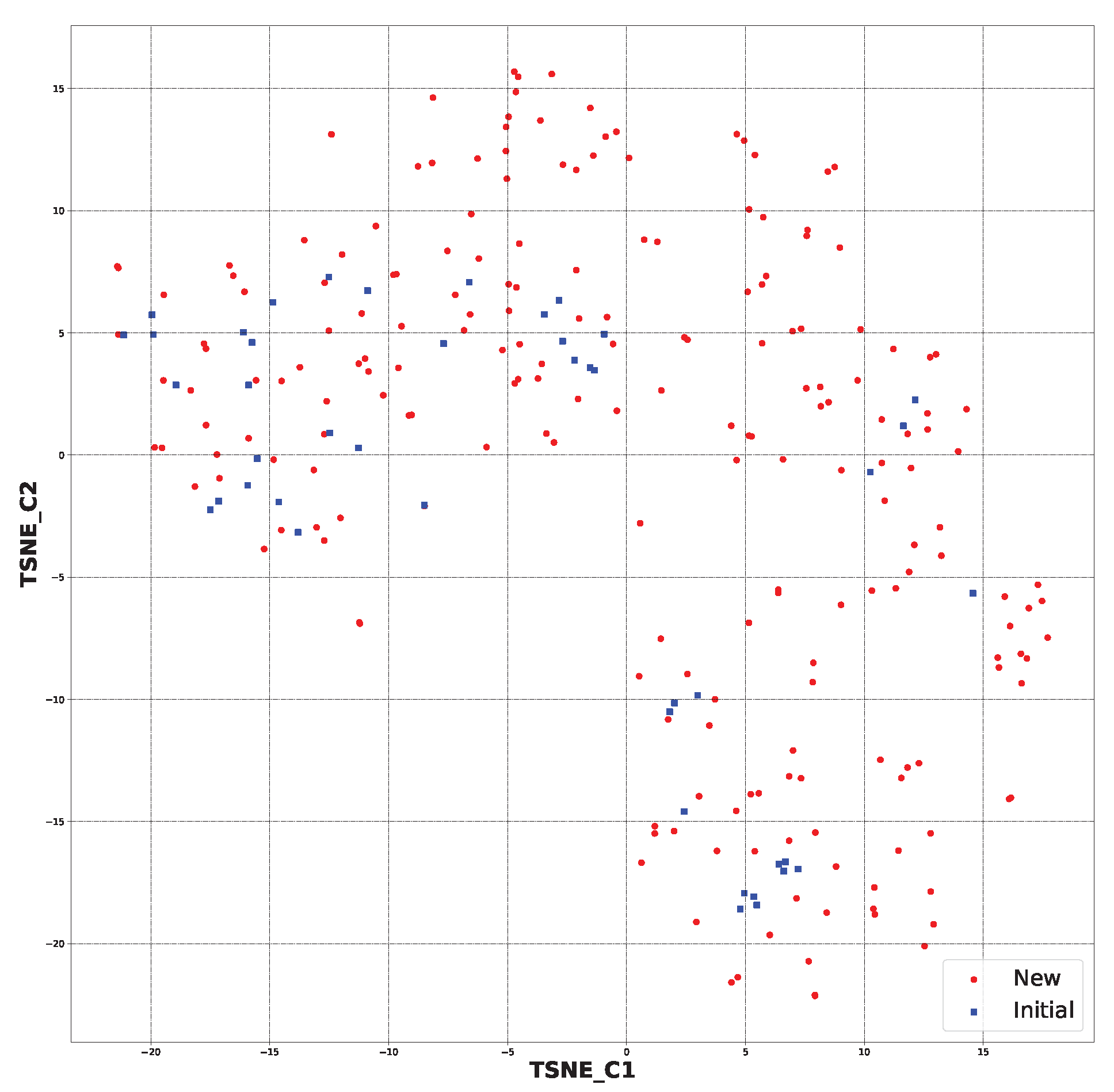
2.1. New Structure Generation
2.2. Cytoprotective Activity Prediction
- The method for predictive model construction based on the approaches listed in Section 3.3;
- The correlation threshold—meaning the correlation between molecular descriptors and measured cytoprotective activity (target feature);
- The number of features (molecular descriptors)—this is closely related to the correlation threshold; the higher the correlation threshold, the fewer features can be used to form a model;
- Standardization of features—when set to “True”, features are standardized; when set to “False”, features are not standardized;
- The training R score indicates how well a model predicts cytoprotective activity (the correlation coefficient between real and predicted values) based on the data seen during training;
- The testing R score indicates how well a model predicts cytoprotective activity (the correlation coefficient between real and predicted values) using data that were not present during training;
- The mean squared error (MSE) displays both the estimator’s bias (accuracy), which is how much its expected value systematically differs from the true value, and the estimator’s variance (precision), which shows how much it fluctuates around its expected value owing to sampling variability [25];
- The mean absolute error (MAE) indicates the average variation between the significant values in the dataset and the projected values in the same dataset [26].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Correlation Threshold | Number of Features | Standardization of Features | Training R Score | Testing R Score | Mean Squared Error | Mean Absolute Error |
|---|---|---|---|---|---|---|---|
| DT [27] | 0.39 | 2 | False | 0.91 | 0.80 | 142.31 | 6.77 |
| RF-12 [28] | 0.39 | 2 | False | 0.85 | 0.75 | 208.96 | 10.54 |
| RF-6 [28] | 0.39 | 2 | False | 0.84 | 0.73 | 226.73 | 10.85 |
| KNN [29] | 0.39 | 2 | True | 0.50 | 0.93 | 975.34 | 20.82 |
| MLR [30] | 0.34 | 9 | False | 0.55 | 0.37 | 530.98 | 16.57 |
| SVR [32] | 0.25 | 46 | True | 0.80 | 0.00 | 1.28 × | 1.84 × |
2.3. Structures for the Experimental Verification
2.4. Additional Analysis of AI-Generated Structures
2.5. Experimental Results
3. Materials and Methods
3.1. Initial Structures
3.2. New Structure Generation
3.3. Cytoprotective Activity Prediction
3.4. Structures for the Experimental Verification
3.5. Additional Analysis of AI-Generated Structures
3.6. Experiment Description
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ROS | reactive oxygen species |
| AI | artificial intelligence |
| SMILES | simplified molecular input line entry specification |
| SELFIES | self-referencing embedded strings |
| SYBA | Synthetic Bayesian classifier |
| DT | decision tree |
| RF | random forest |
| KNN | K-nearest neighbors |
| MLR | multiple linear regression |
| SVR | support vector regression |
References
- Nowak, D.; Bachorz, R.A.; Hoffmann, M. Neural Networks in the Design of Molecules with Affinity to Selected Protein Domains. Int. J. Mol. Sci. 2023, 24, 1762. [Google Scholar] [CrossRef] [PubMed]
- Kotsias, P.C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- Bjerrum, E.J.; Threlfall, R. Molecular Generation with Recurrent Neural Networks (RNNs). arXiv 2017, arXiv:1705.04612. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Bjerrum, E.; Sattarov, B. Improving Chemical Autoencoder Latent Space and Molecular De Novo Generation Diversity with Heteroencoders. Biomolecules 2018, 8, 131. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Jasiewicz, B.; Babijczuk, K.; Warżajtis, B.; Rychlewska, U.; Starzyk, J.; Cofta, G.; Mrówczyńska, L. Indole Derivatives Bearing Imidazole, Benzothiazole-2-Thione or Benzoxazole-2-Thione Moieties—Synthesis, Structure and Evaluation of Their Cytoprotective, Antioxidant, Antibacterial and Fungicidal Activities. Molecules 2023, 28, 708. [Google Scholar] [CrossRef]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef]
- Dorababu, A. Indole—a promising pharmacophore in recent antiviral drug discovery. RSC Med. Chem. 2020, 11, 1335–1353. [Google Scholar] [CrossRef]
- Song, F.; Li, Z.; Bian, Y.; Huo, X.; Fang, J.; Shao, L.; Zhou, M. Indole/isatin-containing hybrids as potential antibacterial agents. Arch. Der Pharm. 2020, 353, 2000143. [Google Scholar] [CrossRef]
- Jasiewicz, B.; Kozanecka-Okupnik, W.; Przygodzki, M.; Warżajtis, B.; Rychlewska, U.; Pospieszny, T.; Mrówczyńska, L. Synthesis, antioxidant and cytoprotective activity evaluation of C-3 substituted indole derivatives. Sci. Rep. 2021, 11, 15425. [Google Scholar] [CrossRef]
- Kozanecka-Okupnik, W.; Sierakowska, A.; Berdzik, N.; Kowalczyk, I.; Mrówczyńska, L.; Jasiewicz, B. New triazole-bearing gramine derivatives—synthesis, structural analysis and protective effect against oxidative haemolysis. Nat. Prod. Res. 2020, 36, 3413–3419. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Kozanecka-Okupnik, W.; Jasiewicz, B.; Pospieszny, T.; Jastrząb, R.; Skrobańska, M.; Mrówczyńska, L. Spectroscopy, molecular modeling and anti-oxidant activity studies on novel conjugates containing indole and uracil moiety. J. Mol. Struct. 2018, 1169, 130–137. [Google Scholar] [CrossRef]
- Silveira, C.C.; Mendes, S.R.; Soares, J.R.; Victoria, F.N.; Martinez, D.M.; Savegnago, L. Synthesis and antioxidant activity of new C-3 sulfenyl indoles. Tetrahedron Lett. 2013, 54, 4926–4929. [Google Scholar] [CrossRef]
- Jacobi, H.; Dell, H.D. [On the pharmacodynamics of acemetacin (author’s transl)]. Arzneim. -Forsch. 1980, 30, 1348–1362. [Google Scholar]
- Atterhög, J.H.; Dunér, H.; Pernow, B. Experience with pindolol, a betareceptor blocker, in the treatment of hypertension. Am. J. Med. 1976, 60, 872–876. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Asmar, R.; O’Rourke, M.F.; Safar, M.E. Mechanism(s) of selective systolic blood pressure reduction after a low-dose combination of perindopril/indapamide in hypertensive subjects: Comparison with atenolol. J. Am. Coll. Cardiol. 2004, 43, 92–99. [Google Scholar] [CrossRef]
- Chen, X.; Zhan, P.; Li, D.; De Clercq, E.; Liu, X. Recent Advances in DAPYs and Related Analogues as HIV-1 NNRTIs. Curr. Med. Chem. 2011, 18, 359–376. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorganic Chem. 2019, 89, 103021. [Google Scholar] [CrossRef]
- Swain, M. PuBChemPy. 2017. Available online: https://github.com/mcs07/PubChemPy/ (accessed on 3 February 2023).
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Open Babel Development Team. Open Babel. 3.1.1. Available online: http://openbabel.org/wiki/Main_Page (accessed on 27 March 2023).
- Schluchter, M.D. Mean Square Error. In Wiley StatsRef: Statistics Reference Online, 1st ed.; Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Fürnkranz, J.; Chan, P.K.; Craw, S.; Sammut, C.; Uther, W.; Ratnaparkhi, A.; Jin, X.; Han, J.; Yang, Y.; Morik, K.; et al. Mean Absolute Error. In Encyclopedia of Machine Learning; Sammut, C., Webb, G.I., Eds.; Springer: Boston, MA, USA, 2011; p. 652. [Google Scholar] [CrossRef]
- von Winterfeldt, D.; Edwards, W. Decision Analysis and Behavioral Research; Cambridge University Press: Cambridge, UK, 1986. [Google Scholar]
- Ho, T.K. Random decision forests. In Proceedings of the Proceedings of 3rd international conference on document analysis and recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Fix, E.; Hodges, J.L. Discriminatory Analysis. Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev./Rev. Int. De Stat. 1989, 57, 238. [Google Scholar] [CrossRef]
- Jobson, J.D. Multiple Linear Regression. In Applied Multivariate Data Analysis; Series Title: Springer Texts in Statistics; Springer: New York, NY, USA, 1991; pp. 219–398. [Google Scholar] [CrossRef]
- Everitt, B.; Skrondal, A. The Cambridge dictionary of statistics, 4th ed.; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Awad, M.; Khanna, R. Support Vector Regression. In Efficient Learning Machines; Apress: New York, NY, USA, 2015; pp. 67–80. [Google Scholar] [CrossRef]
- Mordred Descriptor List. Available online: https://mordred-descriptor.github.io/documentation/master/descriptors.html (accessed on 28 March 2023).
- Roy, K.; Ghosh, G. QSTR with Extended Topochemical Atom Indices. 2. Fish Toxicity of Substituted Benzenes. J. Chem. Inf. Comput. Sci. 2004, 44, 559–567. [Google Scholar] [CrossRef]
- Roy, K.; Das, R. On some novel extended topochemical atom (ETA) parameters for effective encoding of chemical information and modelling of fundamental physicochemical properties. SAR QSAR Environ. Res. 2011, 22, 451–472. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Voršilák, M.; Kolář, M.; Čmelo, I.; Svozil, D. SYBA: Bayesian estimation of synthetic accessibility of organic compounds. J. Cheminform. 2020, 12, 35. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Berdzik, N.; Jasiewicz, B.; Ostrowski, K.; Sierakowska, A.; Slaużys, M.; Nowak, D.; Mrówczyńska, L. Novel gramine-based bioconjugates obtained by click chemistry as cytoprotective compounds and potent antibacterial and antifungal agents. Unpublished.
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Aspuru-Guzik, A. Self-Referencing Embedded Strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: https://zenodo.org/record/7415128 (accessed on 12 December 2022).
- Jurafsky, D.; Martin, J.H. Speech and laNguage Processing: An Introduction to Natural Language Processing, Computational Linguistics, and Speech Recognition; Prentice Hall Series in Artificial Intelligence; Prentice Hall: Upper Saddle River, NJ, USA, 2000. [Google Scholar]
- Categorical Cross-Entropy. Available online: https://peltarion.com/knowledge-center/documentation/modeling-view/build-an-ai-model/loss-functions/categorical-crossentropy (accessed on 21 March 2022).
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Consonni, V.; Todeschini, R. Molecular Descriptors. In Handbook of Computational Chemistry; Leszczynski, J., Ed.; Springer: Dordrecht, The Netherlands, 2016; pp. 1–29. [Google Scholar] [CrossRef]
- Freedman, D.; Pisani, R.; Purves, R. Statistics: Fourth International Student Edition; WW Norton & Company: New York, NY, USA, 2007. [Google Scholar]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry: Miniperspective. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Selected Concepts and Investigations in Compound Classification, Molecular Descriptor Analysis, and Virtual Screening. J. Chem. Inf. Comput. Sci. 2001, 41, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mrówczyńska, L.; Hägerstrand, H. Platelet-activating factor interaction with the human erythrocyte membrane. J. Biochem. Mol. Toxicol. 2009, 23, 345–348. [Google Scholar] [CrossRef] [PubMed]





| Initial structures’ images 1 | ||
 |  |  |
| Newly generated structures’ images 1 | ||
   |    |    |
| Tested structures’ images 1 | ||
 |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowak, D.; Babijczuk, K.; Jaya, L.O.I.; Bachorz, R.A.; Mrówczyńska, L.; Jasiewicz, B.; Hoffmann, M. Artificial Intelligence in Decrypting Cytoprotective Activity under Oxidative Stress from Molecular Structure. Int. J. Mol. Sci. 2023, 24, 11349. https://doi.org/10.3390/ijms241411349
Nowak D, Babijczuk K, Jaya LOI, Bachorz RA, Mrówczyńska L, Jasiewicz B, Hoffmann M. Artificial Intelligence in Decrypting Cytoprotective Activity under Oxidative Stress from Molecular Structure. International Journal of Molecular Sciences. 2023; 24(14):11349. https://doi.org/10.3390/ijms241411349
Chicago/Turabian StyleNowak, Damian, Karolina Babijczuk, La Ode Irman Jaya, Rafał Adam Bachorz, Lucyna Mrówczyńska, Beata Jasiewicz, and Marcin Hoffmann. 2023. "Artificial Intelligence in Decrypting Cytoprotective Activity under Oxidative Stress from Molecular Structure" International Journal of Molecular Sciences 24, no. 14: 11349. https://doi.org/10.3390/ijms241411349
APA StyleNowak, D., Babijczuk, K., Jaya, L. O. I., Bachorz, R. A., Mrówczyńska, L., Jasiewicz, B., & Hoffmann, M. (2023). Artificial Intelligence in Decrypting Cytoprotective Activity under Oxidative Stress from Molecular Structure. International Journal of Molecular Sciences, 24(14), 11349. https://doi.org/10.3390/ijms241411349