
Screening for Novel Inhibitors of Amyloid Beta Aggregation and Toxicity as Potential Drugs for Alzheimer’s Disease

 ,
,
Abstract
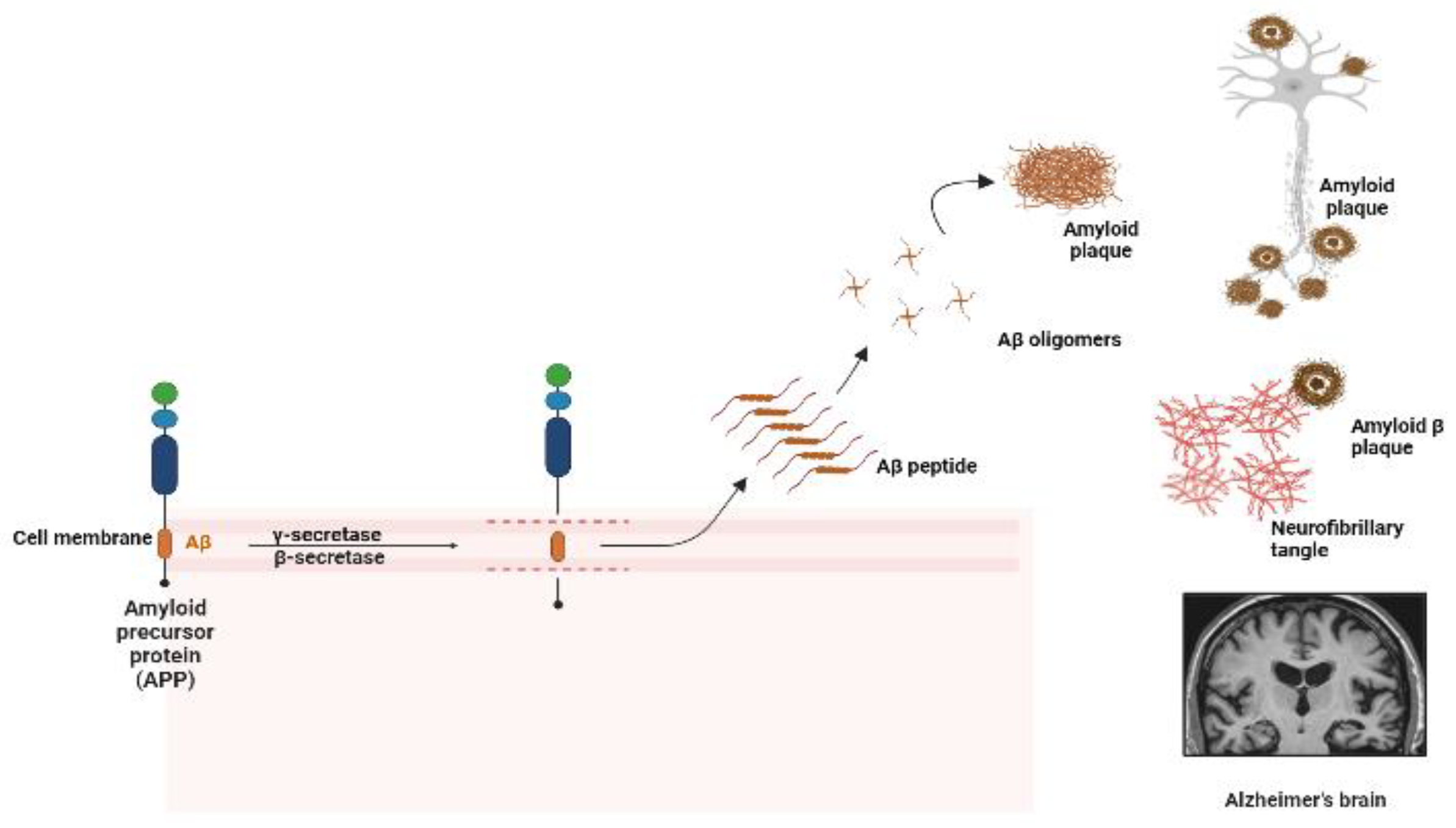
1. Introduction
2. Results
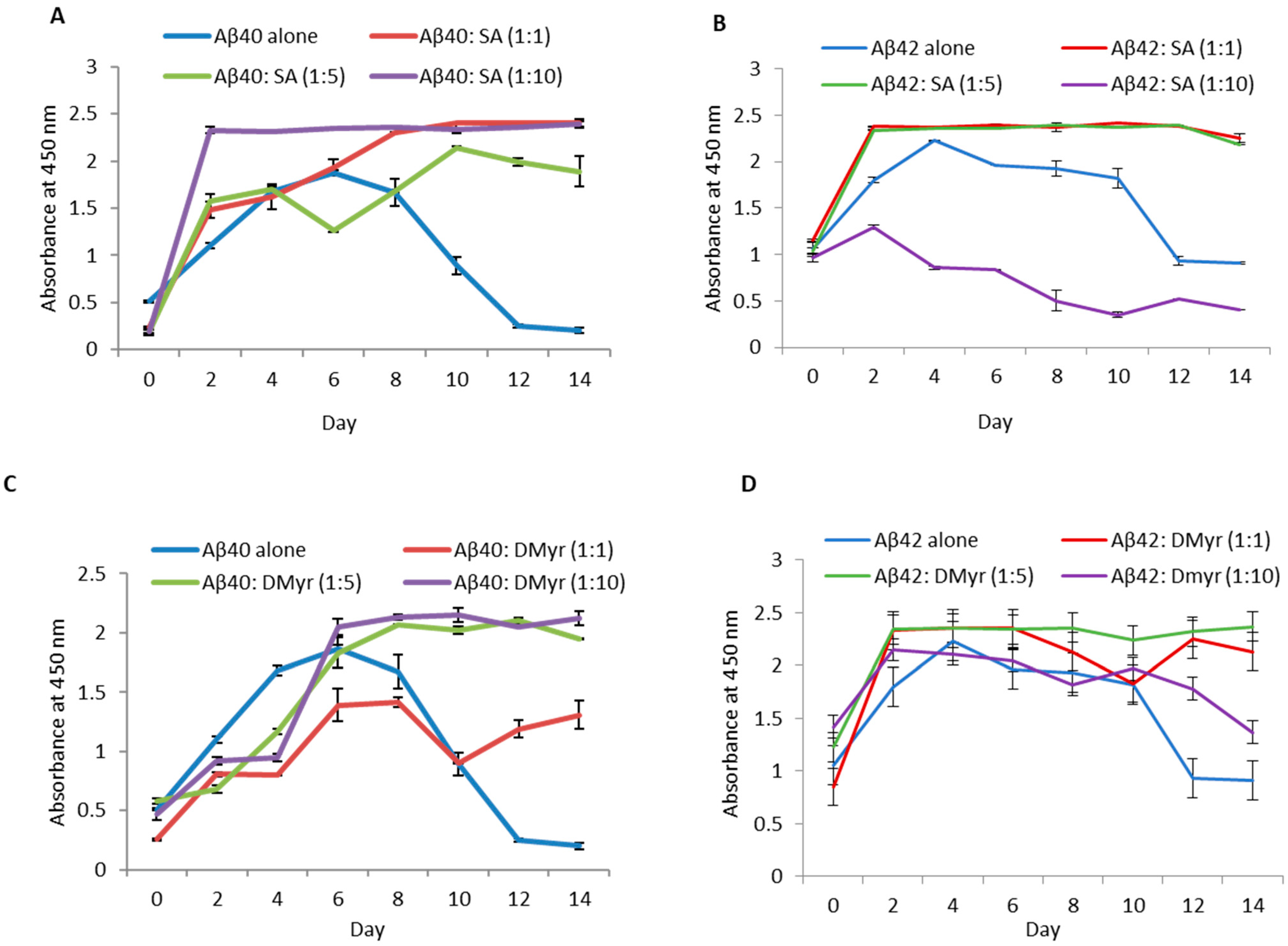
2.1. SA and DMyr, but Not Gn Rb1, Inhibit the Aggregation of Aβ40 and Aβ42 In Vitro
2.2. SA and DMyr Disaggregate Fibrils of Aβ40 and Aβ42
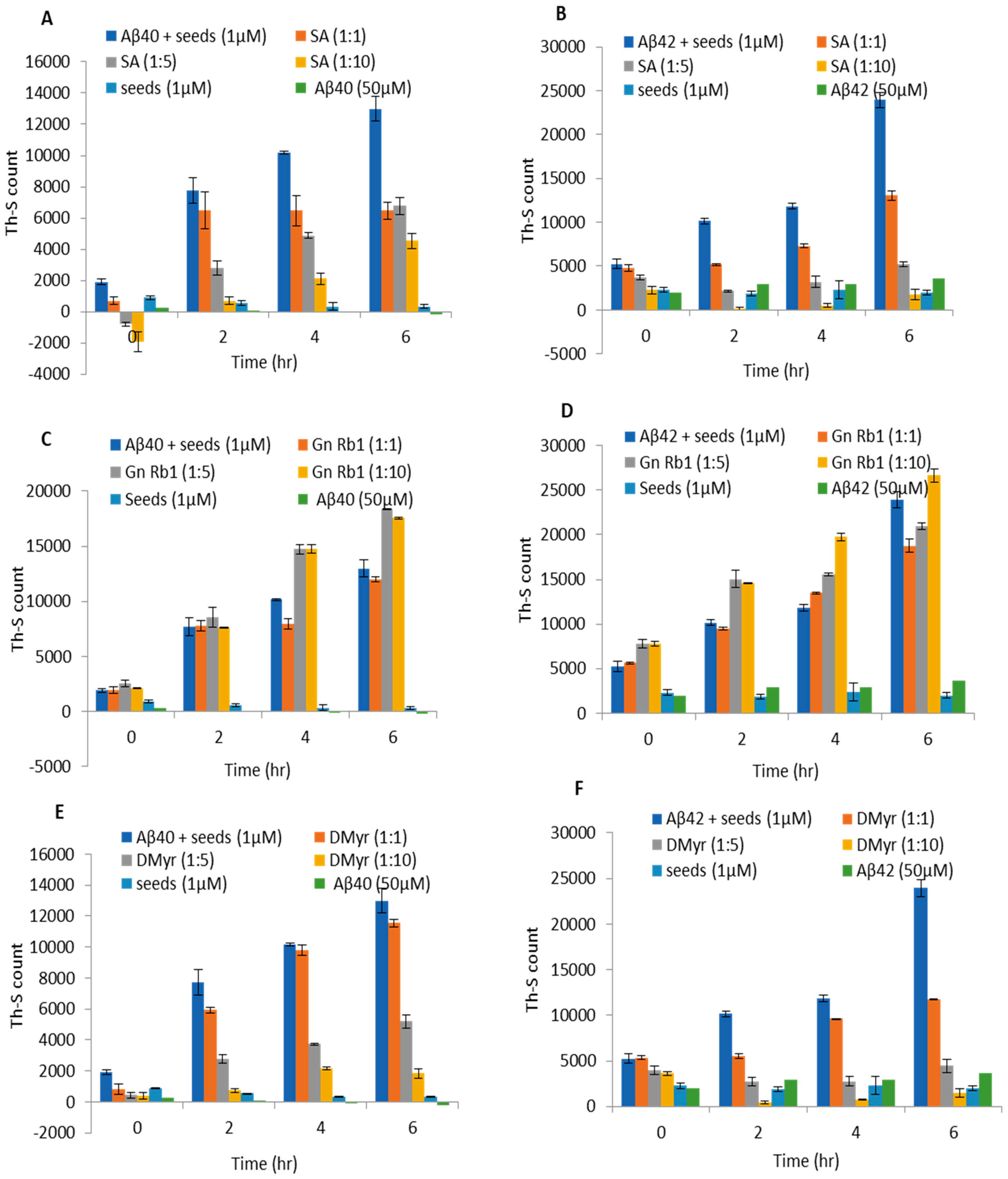
2.3. SA and DMyr Block the Seeded Aggregation of Aβ40 and Aβ42 Monomers
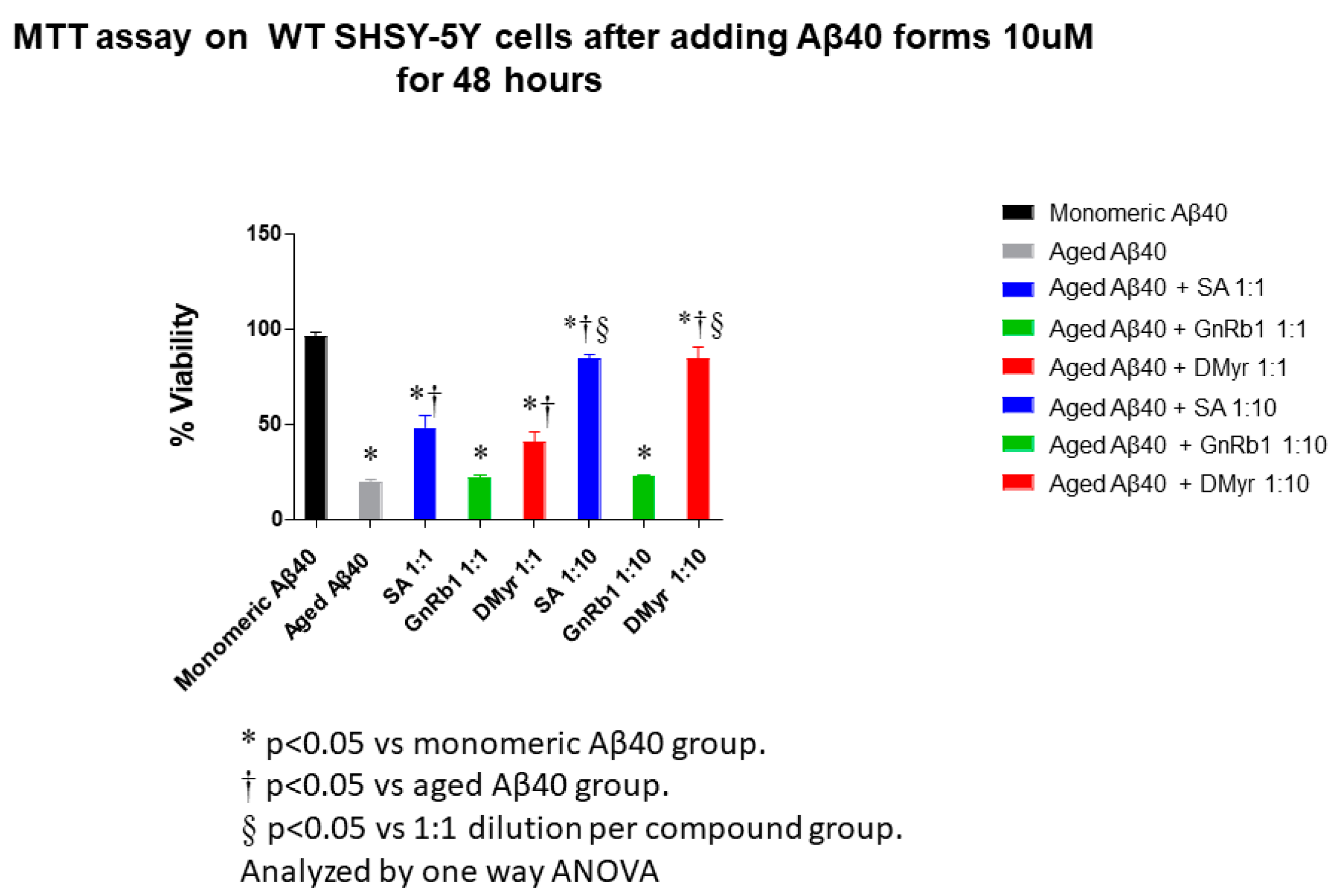
2.4. SA and DMyr Inhibit the Aβ40- and Aβ42-Induced Cytotoxicity in SH-SY5Y Cells
3. Discussion
4. Methods and Materials
4.1. Aggregation of Aβ40 and Aβ42 In Vitro
4.2. Thioflavin-S (Th-S) Assay
4.3. Aβ40 and Aβ42 Disaggregation Assay
4.4. Seeding Polymerization Assay
4.5. Transmission Electron Microscopy (TEM)
4.6. ELISA for Measuring Aβ40 and Aβ42 Early Aggregates
4.7. Culture of SHSY-5Y Human Neuroblastoma Cell Lines
4.8. Measurement of Cell Viability
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| Aβ | Amyloid beta |
| APP | Amyloid precursor protein |
| SA | Salvianolic acid B |
| Gn Rb1 | Ginsenoside Rb1 |
| DMyr | Dihydromyricetin |
| Myr | Myricetin |
| Th-S | Thioflavin-S |
| ELISA | Enzyme-linked immunosorbent assay |
| TEM | Transmission electron microscopy |
References
- Korolev, I.O. Alzheimer’s Disease: A Clinical and Basic Science Review. Med. Student Res. J. 2014, 4, 24–33. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Nisha, C.M.; Silakari, C.; Sharma, I.; Anusha, K.; Gupta, N.; Nair, P.; Tripathi, T.; Kumar, A. Current and novel therapeutic molecules and targets in Alzheimer’s disease. J. Formos. Med. Assoc. 2016, 115, 3–10. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; López, E.S.; García, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2015, 2016, 8501693. [Google Scholar] [CrossRef] [PubMed]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s disease and some therapeutic approaches targeting Aβby using several synthetic and herbal compounds. Oxidative Med. Cell. Longev. 2015, 2016, 7361613. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.; Daggett, V. Amyloid-β Oligomers: Multiple Moving Targets. Biophysica 2022, 2, 91–110. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β (1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.P.; Deverin, S.P.; Inouye, H.; El-Agnaf, O.M.; Teeter, M.M.; Kirschner, D.A. Assemblies of Alzheimer’s peptides Aβ25–35 and Aβ31–35: Reverse-turn conformation and side-chain interactions revealed by X-ray diffraction. J. Struct. Biol. 2003, 141, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Allsop, D.; Swanson, L.; Moore, S.; Davies, Y.; York, A.; El-Agnaf, O.; Soutar, I. Fluorescence Anisotropy: A Method for Early Detection of Alzheimer β-Peptide (Aβ) Aggregation. Biochem. Biophys. Res. Commun. 2001, 285, 58–63. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Multifunction of myricetin on Aβ: Neuroprotection via a conformational change of Aβ and reduction of Aβ via the interference of secretases. J. Neurosci. Res. 2008, 86, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Xin, Y.; Li, Y.; Xu, F.; Xi, X.; Guo, H.; Cui, X.; Cao, H.; Zhang, X.; Han, C. Ginsenosides: A Potential Neuroprotective Agent. BioMed Res. Int. 2018, 2018, 8174345. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, S.; Yu, H.; Duan, S.; Zhao, J. Baicalin and ginsenoside Rb1 promote the proliferation and differentiation of neural stem cells in Alzheimer’s disease model rats. Brain Res. 2018, 1678, 187–194. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhang, Z.; Bi, P.; Qi, Z.; Zhang, C. Anti-neuroinflammation effect of ginsenoside Rbl in a rat model of Alzheimer disease. Neurosci. Lett. 2011, 487, 70–72. [Google Scholar] [CrossRef]
- Lee, Y.W.; Kim, D.H.; Jeon, S.J.; Park, S.J.; Kim, J.M.; Jung, J.M.; Lee, H.E.; Gil Bae, S.; Oh, H.K.; Son, K.H.H.; et al. Neuroprotective effects of salvianolic acid B on an Aβ25–35 peptide-induced mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2013, 704, 70–77. [Google Scholar] [CrossRef]
- Tang, Y.; Huang, D.; Zhang, M.-H.; Zhang, W.-S.; Tang, Y.-X.; Shi, Z.-X.; Deng, L.; Zhou, D.-H.; Lu, X.-Y. Salvianolic acid B inhibits Aβ generation by modulating BACE1 activity in SH-SY5Y-APPsw cells. Nutrients 2016, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Menon, S.A.; Khair, S.B.A.; Lu, J.-H.; Safieh-Garabedian, B.; Al-Hayani, A.A.; Eliezer, D.; Li, M.; et al. Ginsenoside Rb1 inhibits fibrillation and toxicity of alpha-synuclein and disaggregates preformed fibrils. Neurobiol. Dis. 2015, 74, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PLoS Pathog. 2014, 2013, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s disease: Are they effective? Pharm. Ther. 2010, 35, 208–211. [Google Scholar]
- Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.; Twyman, L.J.; Allsop, D. New evidence that the Alzheimer β-amyloid peptide does not spontaneously form free radicals: An ESR study using a series of spin-traps. Free Radic. Biol. Med. 2001, 30, 1154–1162. [Google Scholar] [CrossRef]
- Lansdall, C.J. An effective treatment for Alzheimer’s disease must consider both amyloid and tau. Biosci. Horiz. 2014, 7, hzu002. [Google Scholar] [CrossRef]
- Nie, Q.; Du, X.-G.; Geng, M.-Y. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef]
- Moore, S.A.; Huckerby, T.N.; Gibson, G.L.; Fullwood, N.J.; Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.A.; Allsop, D. Both the d-(+) and l-(−) Enantiomers of Nicotine Inhibit Aβ Aggregation and Cytotoxicity. Biochemistry 2004, 43, 819–826. [Google Scholar] [CrossRef]
- Porzoor, A.; Alford, B.; Hügel, H.M.; Grando, D.; Caine, J.; Macreadie, I. Anti-Amyloidogenic Properties of Some Phenolic Compounds. Biomolecules 2015, 5, 505–527. [Google Scholar] [CrossRef]
- Durairajan, S.S.K.; Yuan, Q.; Xie, L.; Chan, W.-S.; Kum, W.-F.; Koo, I.; Liu, C.; Song, Y.-Q.; Huang, J.-D.; Klein, W.L.; et al. Salvianolic acid B inhibits Aβ fibril formation and disaggregates preformed fibrils and protects against Aβ-induced cytotoxicty. Neurochem. Int. 2008, 52, 741–750. [Google Scholar] [CrossRef]
- Tan, F.H.P.; Ting, A.C.J.; Leow, B.G.; Najimudin, N.; Watanabe, N.; Azzam, G. Alleviatory effects of Danshen, Salvianolic acid A and Salvianolic acid B on PC12 neuronal cells and Drosophila melanogaster model of Alzheimer’s disease. J. Ethnopharmacol. 2021, 279, 114389. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Park, S.; Shin, J.; Lee, J.-S.; Kim, H.Y.; Han, G.; Kim, Y. Investigation of memory-enhancing botanical mixture and their isolated compounds for inhibition of amyloid-β and tau aggregation. Appl. Biol. Chem. 2020, 63, 22. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.; Chung, D.S.; Benedek, G.B.; A Kirschner, D.; Teplow, D.B. On the nucleation and growth of amyloid beta-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. USA 1996, 93, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Gao, S.; Wang, T.; Shen, Y.; Yang, W.; Li, Y.; Hu, H. Ginsenoside Rb1 improves learning and memory ability through its anti-inflammatory effect in Aβ1-40 induced Alzheimer’s disease of rats. Am. J. Transl. Res. 2019, 11, 2955–2968. [Google Scholar]
- Cho, I.-H. Effects of Panax ginseng in Neurodegenerative Diseases. J. Ginseng Res. 2012, 36, 342–353. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.-I.; Masuda, Y.; Takegoshi, K.; Irie, K. Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhao, W.; Sang, J.; Wang, W.; Wei, W.; Wang, Y.; Zhao, F.; Lu, F.; Liu, F. Inhibitory Effect of a Flavonoid Dihydromyricetin against Aβ40 Amyloidogenesis and Its Associated Cytotoxicity. ACS Chem. Neurosci. 2019, 10, 4696–4703. [Google Scholar] [CrossRef]
- Stefanescu, R.; Stanciu, G.D.; Luca, A.; Paduraru, L.; Tamba, B.-I. Secondary metabolites from plants possessing inhibitory properties against beta-amyloid aggregation as revealed by thioflavin-T assay and correlations with investigations on transgenic mouse models of Alzheimer’s disease. Biomolecules 2020, 10, 870. [Google Scholar] [CrossRef]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.-I.; Nishijo, H.; et al. Phenolic Compounds Prevent Amyloid β-Protein Oligomerization and Synaptic Dysfunction by Site-specific. J. Biol. Chem. 2012, 287, 14631–14643. [Google Scholar] [CrossRef]
- Liu, M.; Guo, H.; Li, Z.; Zhang, C.; Zhang, X.; Cui, Q.; Tian, J. Molecular Level Insight Into the Benefit of Myricetin and Dihydromyricetin Uptake in Patients With Alzheimer’s Diseases. Front. Aging Neurosci. 2020, 12, 601603. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Abbreviation | Previous Studies | New Findings |
|---|---|---|---|
| Salvianolic acid B | SA |
|
|
| Dihydromyricetin | DMyr |
|
|
| Ginsenoside Rb1 | Gn Rb1 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharari, S.; Vaikath, N.N.; Tsakou, M.; Ghanem, S.S.; Vekrellis, K. Screening for Novel Inhibitors of Amyloid Beta Aggregation and Toxicity as Potential Drugs for Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 11326. https://doi.org/10.3390/ijms241411326
Sharari S, Vaikath NN, Tsakou M, Ghanem SS, Vekrellis K. Screening for Novel Inhibitors of Amyloid Beta Aggregation and Toxicity as Potential Drugs for Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(14):11326. https://doi.org/10.3390/ijms241411326
Chicago/Turabian StyleSharari, Sanaa, Nishant N. Vaikath, Magdalini Tsakou, Simona S. Ghanem, and Kostas Vekrellis. 2023. "Screening for Novel Inhibitors of Amyloid Beta Aggregation and Toxicity as Potential Drugs for Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 14: 11326. https://doi.org/10.3390/ijms241411326
APA StyleSharari, S., Vaikath, N. N., Tsakou, M., Ghanem, S. S., & Vekrellis, K. (2023). Screening for Novel Inhibitors of Amyloid Beta Aggregation and Toxicity as Potential Drugs for Alzheimer’s Disease. International Journal of Molecular Sciences, 24(14), 11326. https://doi.org/10.3390/ijms241411326