

Age, Origin and Functional Study of the Prevalent LDLR Mutation Causing Familial Hypercholesterolaemia in Gran Canaria
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
2.1. Demographic, Clinical and Genetic Characterisation
2.2. Age of the p.(Tyr400_Phe402del) Mutation

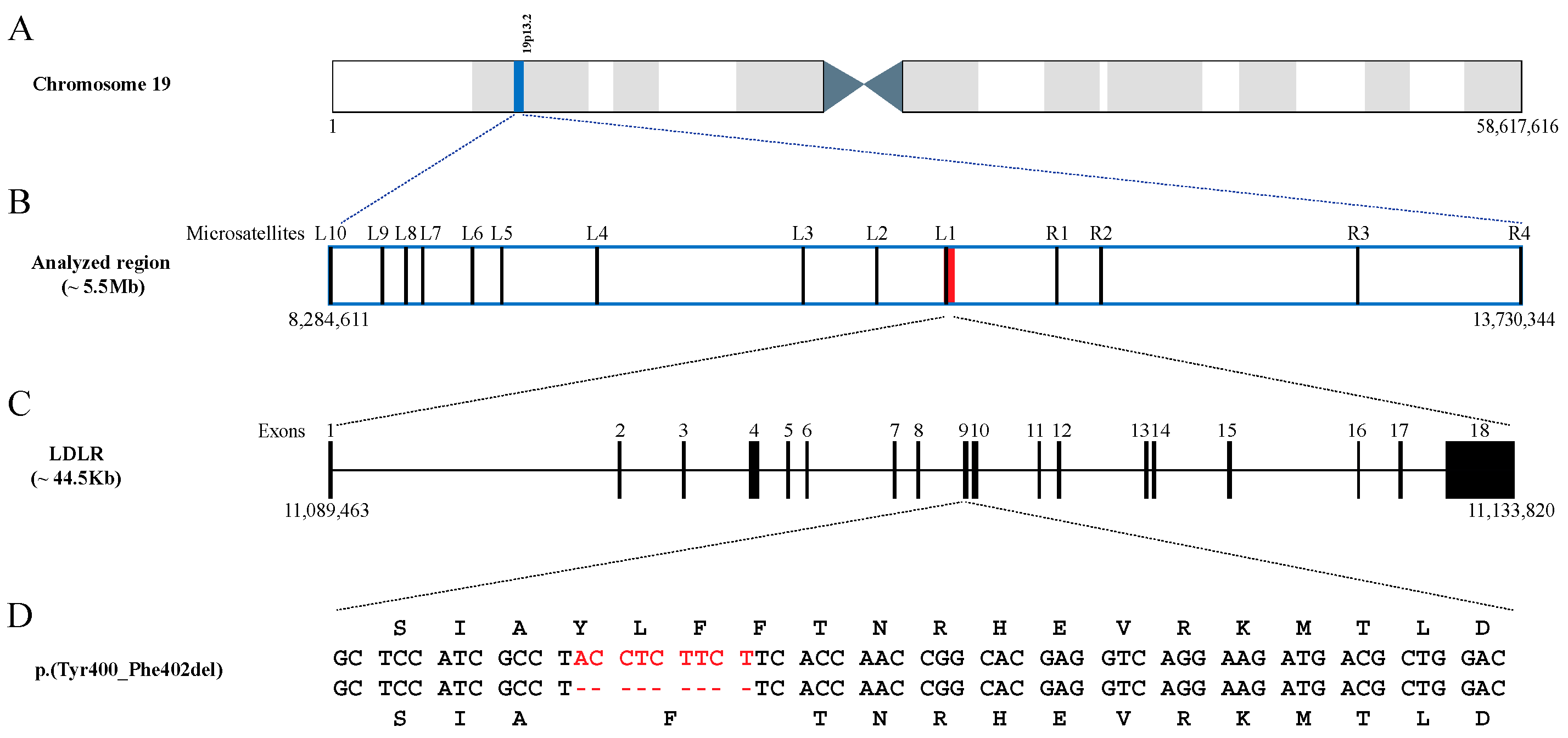{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microsatellite Markers (Distance (cM) from the Mutation) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L10 (4.69) | L9 (4.19) | L8 (4.04) | L7 (3.82) | L6 (3.20) | L5 (2.89) | L4 (1.85) | L3 (0.38) | L2 (0.05) | L1 (0.01) | p.(Tyr400_Phe402del) | R1 (0.99) | R2 (1.31) | R3 (2.16) | R4 (4.17) | ||
| Haplotypes (frequency) | 1 (54.8%) | 327 | 193 | 241 | 225 | 245 | 225 | 226 | 143 | 163 | 119 | 374 | 159 | 256 | 205 | |
| 2 (3.2%) | 329 | 193 | 243 | 225 | 245 | 225 | 226 | 143 | 163 | 119 | 374 | 159 | 256 | 205 | ||
| 3 (16.1%) | 327 | 193 | 241 | 225 | 245 | 225 | 226 | 143 | 163 | 119 | 374 | 159 | 260 | 205 | ||
| 4 (12.9%) | 335 | 203 | 241 | 225 | 245 | 225 | 226 | 143 | 163 | 119 | 374 | 159 | 256 | 205 | ||
| 5 (3.2%) | 335 | 203 | 241 | 225 | 245 | 225 | 226 | 143 | 163 | 119 | 374 | 159 | 256 | 207 | ||
| 6 (3.2%) | 327 | 193 | 241 | 225 | 245 | 225 | 224 | 143 | 163 | 119 | 374 | 159 | 260 | 205 | ||
| 7 (3.2%) | 335 | 193 | 241 | 225 | 245 | 225 | 226 | 143 | 163 | 119 | 374 | 159 | 256 | 205 | ||
| 8 (3.2%) | 339 | 203 | 241 | 239 | 245 | 225 | 226 | 143 | 163 | 119 | 388 | 159 | 260 | 205 | ||
| Generations (years) | CI-lower | CI-upper | ||||||||||||||
| Assuming a ‘correlated’ genealogy | 15.5 [387] | 4.4 [110] | 62.9 (1572) | |||||||||||||

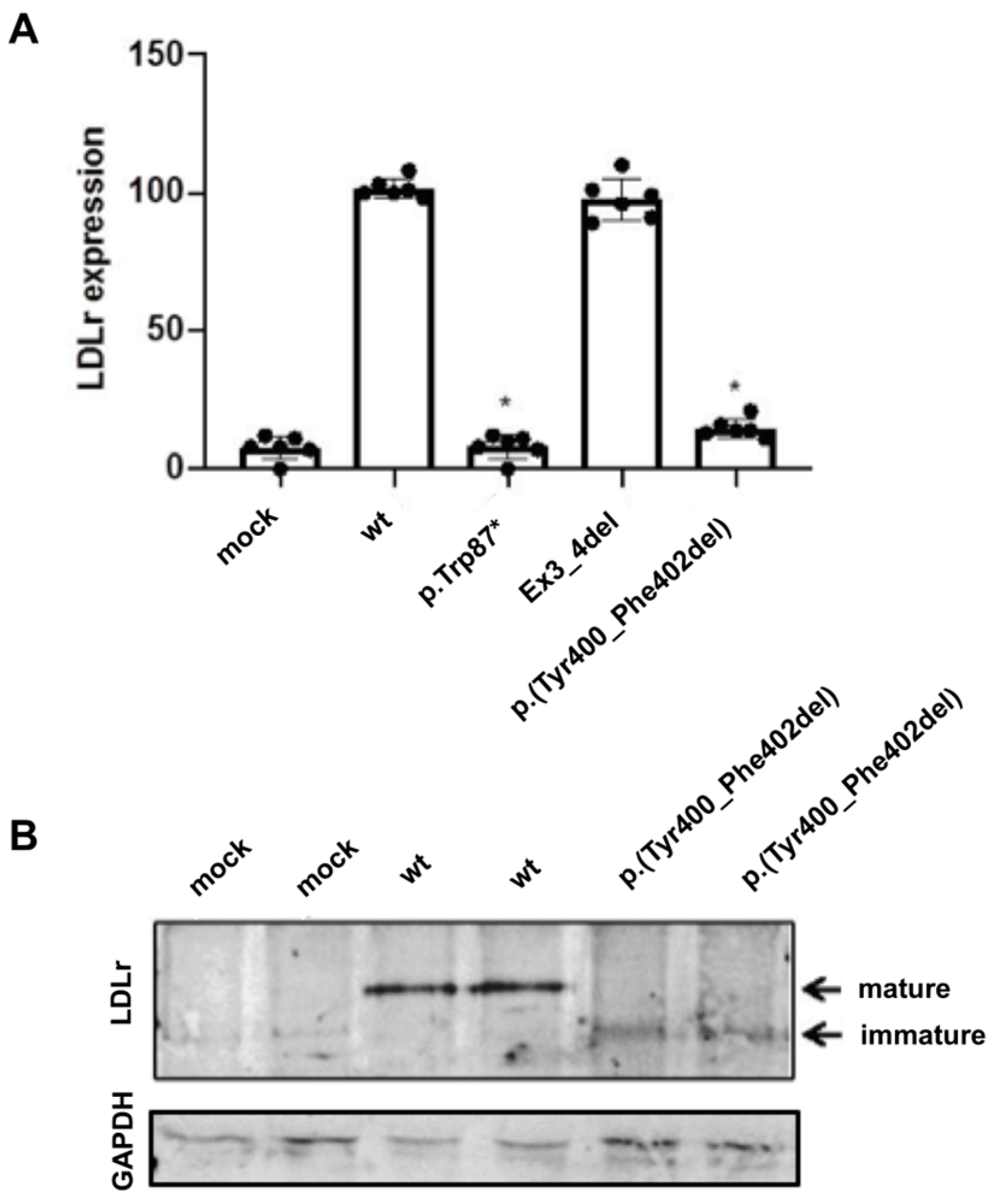
2.3. Expression of the p.(Tyr400_Phe402del) LDLR Variant in CHO-ldlA7 Cells
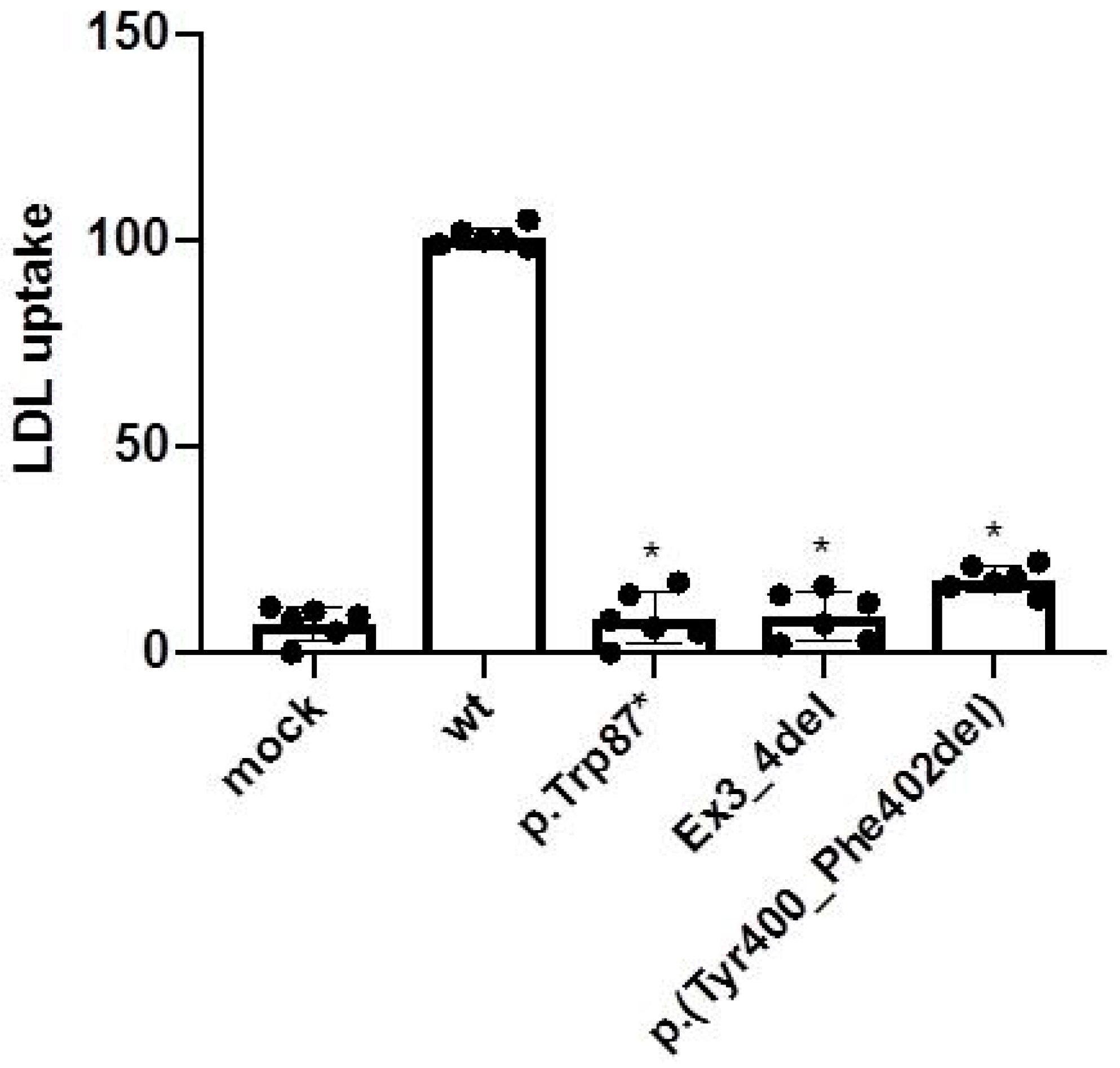
2.4. LDL Uptake Activity of the p.(Tyr400_Phe402del) LDLR Variant in CHO-ldlA7 Cells
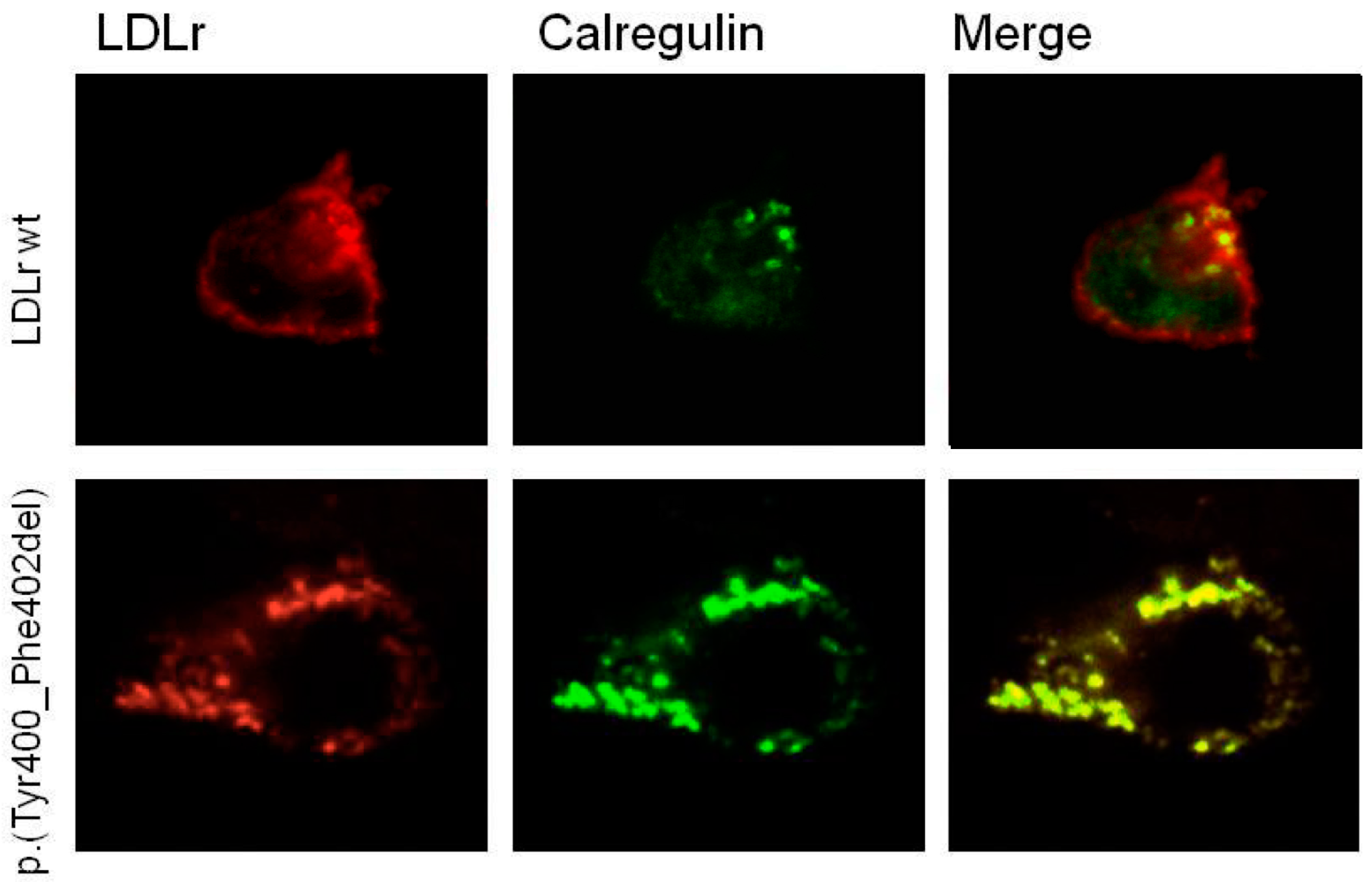
2.5. p.(Tyr400_Phe402del) LDLR Variant Classification by Confocal Microscopy
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Microsatellite Genotyping
4.3. Genetic Characterisation
4.4. Estimation of the Age of the Variant
4.5. Functional Characterisation of the Variant
4.5.1. Cloning of LDLR Variant
4.5.2. CHO-ldlA7 Cell Culture and Transfection
4.5.3. Immunodetection of LDLr by Western Blot
4.5.4. Analysis of LDLR Expression by Fluorescent Activated Cell Sorter (FACS)
4.5.5. Analysis of LDL Uptake by FACS
4.5.6. Confocal Laser Scanning Microscopy
4.5.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. Available online: https://academic.oup.com/eurheartj/article/34/45/3478/435928 (accessed on 13 January 2022). [CrossRef] [PubMed]
- Goldstein, J.L.; Schrott, H.G.; Hazzard, W.R.; Bierman, E.L.; Motulsky, A.G. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Investig. 1973, 52, 1544–1568. Available online: https://pubmed.ncbi.nlm.nih.gov/4718953/ (accessed on 13 January 2022). [CrossRef]
- Bruikman, C.S.; Hovingh, G.K.; Kastelein, J.J.P. Molecular basis of familial hypercholesterolemia. Curr. Opin. Cardiol. 2017, 32, 262–266. Available online: https://pubmed.ncbi.nlm.nih.gov/28169949/ (accessed on 13 January 2022). [CrossRef] [PubMed]
- Vrablik, M.; Tichý, L.; Freiberger, T.; Blaha, V.; Satny, M.; Hubacek, J.A. Genetics of familial hypercholesterolemia: New insights. Front. Genet. 2020, 11, 574474. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Raal, F.J.; Hegele, R.A.; Al-Rasadi, K.; Arca, M.; Averna, M.; Bruckert, E.; Freiberger, T.; Gaudet, D.; Harada-Shiba, M.; et al. 2023 update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: New treatments and clinical guidance. Eur. Heart J. 2023, 44, ehad197. Available online: https://pubmed.ncbi.nlm.nih.gov/37130090/ (accessed on 13 June 2023).
- Kotze, M.J.; Langenhoven, E.; Warnich, L.; Plessis, L.D.U.; Retief, A.E. The molecular basis and diagnosis of familial hypercholesterolaemia in South African Afrikaners. Ann. Hum. Genet. 1991, 55, 115–121. Available online: https://pubmed.ncbi.nlm.nih.gov/1952806/ (accessed on 13 January 2022). [CrossRef]
- Meiner, V.; Landsberger, D.; Berkman, N.; Reshef, A.; Segal, P.; Seftel, H.C.; van der Westhuyzen, D.R.; Jeenah, M.S.; Coetzee, G.A.; Leitersdorf, E. A common Lithuanian mutation causing familial hypercholesterolemia in Ashkenazi Jews. Am. J. Hum. Genet. 1991, 49, 443. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1683281/ (accessed on 13 January 2022).
- Couture, P.; Morissette, J.; Gaudet, D.; Vohl, M.C.; Gagné, C.; Bergeron, J.; Després, J.P.; Simard, J. Fine mapping of low-density lipoprotein receptor gene by genetic linkage on chromosome 19p13.1-p13.3 and study of the founder effect of four French Canadian low-density lipoprotein receptor gene mutations. Atherosclerosis 1999, 143, 145–151. Available online: https://pubmed.ncbi.nlm.nih.gov/10208489/ (accessed on 13 January 2022). [CrossRef]
- Abifadel, M.; Rabès, J.P.; Jambart, S.; Halaby, G.; Gannagé-Yared, M.H.; Sarkis, A.; Beaino, G.; Varret, M.; Salem, N.; Corbani, S.; et al. The molecular basis of familial hypercholesterolemia in Lebanon: Spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum. Mutat. 2009, 30, E682–E691. Available online: https://pubmed.ncbi.nlm.nih.gov/19319977/ (accessed on 23 February 2022). [CrossRef]
- Lahtinen, A.M.; Havulinna, A.S.; Jula, A.; Salomaa, V.; Kontula, K. Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis 2015, 238, 64–69. Available online: https://pubmed.ncbi.nlm.nih.gov/25437892/ (accessed on 23 February 2022). [CrossRef]
- Sánchez-Hernández, R.M.; Tugores, A.; Nóvoa, F.J.; Brito-Casillas, Y.; Expósito-Montesdeoca, A.B.; Garay, P.; Bea, A.M.; Riaño, M.; Pocovi, M.; Civeira, F.; et al. The island of Gran Canaria: A genetic isolate for familial hypercholesterolemia. J. Clin. Lipidol. 2019, 13, 618–626. [Google Scholar] [CrossRef]
- Palacios, L.; Grandoso, L.; Cuevas, N.; Olano-Martín, E.; Martinez, A.; Tejedor, D.; Stef, M. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis 2012, 221, 137–142. Available online: https://pubmed.ncbi.nlm.nih.gov/22244043/ (accessed on 23 February 2022). [CrossRef] [PubMed]
- Sánchez-Hernández, R.M.; González-Lleó, A.M.; Tugores, A.; Brito-Casillas, Y.; Civeira, F.; Boronat, M.; Wägner, A. Familial hypercholesterolemia in Gran Canaria: Founder mutation effect and high frequency of diabetes. Clin. Investig. Arterioscler. 2021, 33, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Mozas, P.; Castillo, S.; Tejedor, D.; Reyes, G.; Alonso, R.; Franco, M.; Saenz, P.; Fuentes, F.; Almagro, F.; Mata, P.; et al. Molecular characterization of familial hypercholesterolemia in Spain: Identification of 39 novel and 77 recurrent mutations in LDLR. Hum. Mutat. 2004, 24, 187. Available online: https://pubmed.ncbi.nlm.nih.gov/15241806/ (accessed on 23 February 2022). [CrossRef] [PubMed]
- de Abreu Galindo, J.; Cioranescu, A. Historia de la Conquista de las Siete Islas de Canaria; Goya Ediciones: Santa Cruz de Tenerife, Spain, 1977. [Google Scholar]
- Flores, C.; Larruga, J.M.; González, A.M.; Hernández, M.; Pinto, F.M.; Cabrera, V.M. The origin of the Canary Island aborigines and their contribution to the modern population: A molecular genetics perspective. Curr. Anthropol. 2001, 42, 749–755. Available online: https://www.journals.uchicago.edu/doi/10.1086/323819 (accessed on 14 March 2022). [CrossRef]
- Guillen-Guio, B.; Lorenzo-Salazar, J.M.; Gonzalez-Montelongo, R.; De Usera, A.D.; Marcelino-Rodrıguez, I.; Corrales, A.; de León, A.C.; Alonso, S.; Flores, C. Genomic Analyses of Human European Diversity at the Southwestern Edge: Isolation, African Influence and Disease Associations in the Canary Islands. Mol. Biol. Evol. 2018, 35, 3010–3026. Available online: https://academic.oup.com/mbe/article/35/12/3010/5115937 (accessed on 28 September 2022). [CrossRef]
- Lorente-Arencibia, P.; Garciá-Villarreal, L.; González-Montelongo, R.; Rubio-Rodríguez, L.A.; Flores, C.; Garay-Sánchez, P.; de la Cruz, T.; Santana-Verano, M.; Rodríguez-Esparragón, F.; Benitez-Reyes, J.N.; et al. Wilson Disease Prevalence: Discrepancy between Clinical Records, Registries and Mutation Carrier Frequency. J. Pediatr. Gastroenterol. Nutr. 2022, 74, 192–199. Available online: https://europepmc.org/article/med/34620762 (accessed on 28 September 2022). [CrossRef]
- Peña-Quintana, L.; Scherer, G.; Curbelo-Estévez, M.L.; Jiménez-Acosta, F.; Hartmann, B.; La Roche, F.; Meavilla-Olivas, S.; Pérez-Cerdá, C.; García-Segarra, N.; Giguère, Y.; et al. Tyrosinemia type II: Mutation update, 11 novel mutations and description of 5 independent subjects with a novel founder mutation. Clin. Genet. 2017, 92, 306–317. Available online: https://pubmed.ncbi.nlm.nih.gov/28255985/ (accessed on 28 September 2022). [CrossRef]
- Genin, E.; Tullio-Pelet, A.; Begeot, F.; Lyonnet, S.; Abel, L. Estimating the age of rare disease mutations: The example of Triple-A syndrome. J. Med. Genet. 2004, 41, 445–449. Available online: https://pubmed.ncbi.nlm.nih.gov/15173230/ (accessed on 8 May 2023). [CrossRef]
- Colombo, R. Dating Mutations. In Handbook of Human Molecular Evolution; Wiley: New York, NY, USA, 2008; pp. 138–142. [Google Scholar]
- Gandolfo, L.C.; Bahlo, M.; Speed, T.P. Dating rare mutations from small samples with dense marker data. Genetics 2014, 197, 1315–1327. Available online: https://pubmed.ncbi.nlm.nih.gov/24879464/ (accessed on 8 May 2023). [CrossRef]
- Etxebarria, A.; Palacios, L.; Stef, M.; Tejedor, D.; Uribe, K.B.; Oleaga, A.; Pérez-Sánchez, R.; Cabezas-Cruz, A. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum. Mutat. 2012, 33, 232–243. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/humu.21630 (accessed on 15 October 2022). [CrossRef]
- Rannala, B.; Bertorelle, G. Using linked markers to infer the age of a mutation. Hum. Mutat. 2001, 18, 87–100. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/humu.1158 (accessed on 8 May 2023). [CrossRef] [PubMed]
- Fernández-Armesto, F. The Canary Islands after the Conquest: The Making of a Colonial Society in the Early Sixteenth Century; Clarendon Press: Oxford, UK, 1982; p. 244. [Google Scholar]
- Durst, R.; Colombo, R.; Shpitzen, S.; Avi, L.; Ben Friedlander, Y.; Wexler, R.; Raal, F.J.; Marais, D.A.; Defesche, J.C.; Mandelshtam, M.Y.; et al. Recent origin and spread of a common lithuanian mutation, G197del LDLR, causing familial hypercholesterolemia: Positive selection is not always necessary to account for disease incidence among Ashkenazi Jews. Am. J. Hum. Genet. 2001, 68, 1172–1188. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Pérez, J.; de León Hernández, J.C.; Pérez-Pérez, H.; Mendoza-Grimón, M.D.; Gutierrez-Martinez, A.J.; Hadjigeorgiou, I.; Montón-Álvarez, F.; González-Quereda, L.; Alonso-Jimenez, A.; Suárez-Calvet, X.; et al. Clinical and genetic features of a large homogeneous cohort of oculopharyngeal muscular dystrophy patients from the Canary Islands. Eur. J. Neurol. 2022, 29, 1488–1495. Available online: https://pubmed.ncbi.nlm.nih.gov/35112761/ (accessed on 8 February 2023). [CrossRef]
- Corona, E.; Dudley, J.T.; Butte, A.J. Extreme Evolutionary Disparities Seen in Positive Selection across Seven Complex Diseases. PLoS ONE 2010, 5, e12236. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0012236 (accessed on 8 February 2023). [CrossRef]
- Besseling, J.; Kastelein, J.J.P.; Defesche, J.C.; Hutten, B.A.; Hovingh, G.K. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA 2015, 313, 1029–1036. Available online: https://pubmed.ncbi.nlm.nih.gov/25756439/ (accessed on 8 February 2023). [CrossRef] [PubMed]
- Climent, E.; Pérez-Calahorra, S.; Marco-Benedí, V.; Plana, N.; Sánchez, R.; Ros, E.; Ascaso, J.F.; Puzo, J.; Almagro, F.; Lahoz, C.; et al. Effect of LDL cholesterol, statins and presence of mutations on the prevalence of type 2 diabetes in heterozygous familial hypercholesterolemia. Sci. Rep. 2017, 7, 5596. [Google Scholar] [CrossRef]
- Jeon, H.; Meng, W.; Takagi, J.; Eck, M.J.; Springer, T.A.; Blacklow, S.C. Implications for familial hypercholesterolemia from the structure of the LDL receptor YWTD-EGF domain pair. Nat. Struct. Biol. 2001, 8, 499–504. Available online: https://pubmed.ncbi.nlm.nih.gov/11373616/ (accessed on 10 March 2023). [CrossRef]
- Ellgaard, L.; Molinari, M.; Helenius, A. Setting the standards: Quality control in the secretory pathway. Science 1999, 286, 1882–1888. Available online: https://pubmed.ncbi.nlm.nih.gov/10583943/ (accessed on 10 March 2023). [CrossRef]
- Labuda, D.; Zieđtkiewicz, E.; Labuda, M. The genetic clock and the age of the founder effect in growing populations: A lesson from French Canadians and Ashkenazim. Am. J. Hum. Genet. 1997, 61, 768–771. Available online: https://europepmc.org/articles/PMC1715935 (accessed on 10 March 2023). [CrossRef]
- Miller, S.; Dykes, D.; Polesky, H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC334765/ (accessed on 23 February 2022). [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. Available online: https://pubmed.ncbi.nlm.nih.gov/21565059/ (accessed on 8 May 2023). [CrossRef] [PubMed]
- Rice, W.R. Analyzing tables of statistical test. Evolution 1989, 43, 223–225. Available online: https://pubmed.ncbi.nlm.nih.gov/28568501/ (accessed on 8 May 2023). [CrossRef] [PubMed]
- Etxebarria, A.; Benito-Vicente, A.; Stef, M.; Ostolaza, H.; Palacios, L.; Martin, C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis 2015, 238, 304–312. [Google Scholar] [CrossRef] [PubMed]



| Gender (% females) | 42.3 |
| Age (years) | 45.1 ± 18.4 |
| BMI (Kg/m2) | 25.5 ± 5.2 |
| Tendinous xanthomas (%) | 42.3 |
| ASCVD (%) | 15.4 |
| Total cholesterol (mg/dL) | 385.1 ± 69.2 |
| HDL-c (mg/dL) | 62.2 ± 20.3 |
| LDL-c (mg/dL) | 299.2 ± 60.7 |
| Triglycerides (mg/dL) | 100.2 ± 42 |
| Lipoprotein (a) (mg/dL), median [IQR] | 30 [13.5–85] |
| Locus. | Primer Sequence (5′-3′) Forward | Primer Sequence (5′-3′) Reverse | Label a | Repeat Motif | Genomic Location ‡ | NA | HO * | HE * | |
|---|---|---|---|---|---|---|---|---|---|
| L10 | GAGGCTGAGACGGGAGAATC | TTCCCCAACACACAAAGCAG | 6-FAM | CA | 8,284,611 | 8,284,940 | 13 | 0.833 | 0.825 |
| L9 | CATGCTCAGCTTCCCAAGAC | AGGTGGAGGTTGCAGTGAG | PET | GT | 8,518,249 | 8,518,442 | 6 | 0.712 | 0.680 |
| L8 | GACTTAGAATGGTGCCTGGC | AAAATTAGCTGGGCACGGTG | NED | GT | 8,622,652 | 8,622,894 | 9 | 0.551 | 0.592 |
| L7 | GTTTCTCACGGCTGACTTGG | CACCTGGCCTCACTTGATGT | VIC | AG | 8,694,731 | 8,695,961 | 12 | 0.897 | 0.889 |
| L6 | GGATGAGTGTGCTTTCTACCC | GGCCCCATATGAACCGTTTC | 6-FAM | GT | 8,928,205 | 8,928,445 | 7 | 0.645 | 0.745 |
| L5 | GCTATTTGGGGTCTCTATCAATG | GAAATCGCACAGTATTTGTCTCAC | VIC | CA | 9,067,697 | 9,067,918 | 13 | 0.667 | 0.821 |
| L4 | AGAAGCTAGGACCACAGACG | ATGCACACCTGTAGTCCCAG | NED | TG | 9,501,287 | 9,501,509 | 10 | 0.854 | 0.821 |
| L3 | GGGTCTGAGGATGTTTCTGC | GCAAATATCCACTGCCCTTG | NED | GT | 10,451,997 | 10,452,137 | 9 | 0.658 | 0.670 |
| L2 | GGGTGCTAGGATTTGGGACT | CATTTGGTCTTGCTCCTCTGA | PET | GT | 10,794,316 | 10,794,475 | 9 | 0.444 | 0.445 |
| L1 | AGTGTGGAAGGAAAAGGGAC | CCAATTCTAGATGGGTCG | 6-FAM | ATA | 11,092,150 | 11,092,197 | 6 | 0.256 | 0.246 |
| R1 | TCCAGCAATTGTTCCCATTCTC | TACACAAACATTAGCCGGGC | 6-FAM | TA | 11,609,282 | 11,609,694 | 16 | 0.274 | 0.646 |
| R2 | AGATCGCACCACTGTACTCC | TTCCCGCCTAGTAACGGAC | VIC | CA | 11,815,227 | 11,815,384 | 16 | 0.793 | 0.829 |
| R3 | TCTTCCCATTGCAGTTGTGG | AACACCCTCCCCATGTACAC | PET | GT | 12,988,231 | 12,988,486 | 9 | 0.778 | 0.708 |
| R4 | ATAGGCCAAGACTGTCTAAAACAA | GCCCTAACTGCTGTAAGAGAACT | 6-FAM | CA | 13,730,141 | 13,730,344 | 9 | 0.737 | 0.683 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez, N.M.; Jebari-Benslaiman, S.; Jiménez-Monzón, R.; Benito-Vicente, A.; Brito-Casillas, Y.; Garcés, L.; González-Lleo, A.M.; Tugores, A.; Boronat, M.; Martin, C.; et al. Age, Origin and Functional Study of the Prevalent LDLR Mutation Causing Familial Hypercholesterolaemia in Gran Canaria. Int. J. Mol. Sci. 2023, 24, 11319. https://doi.org/10.3390/ijms241411319
Suárez NM, Jebari-Benslaiman S, Jiménez-Monzón R, Benito-Vicente A, Brito-Casillas Y, Garcés L, González-Lleo AM, Tugores A, Boronat M, Martin C, et al. Age, Origin and Functional Study of the Prevalent LDLR Mutation Causing Familial Hypercholesterolaemia in Gran Canaria. International Journal of Molecular Sciences. 2023; 24(14):11319. https://doi.org/10.3390/ijms241411319
Chicago/Turabian StyleSuárez, Nicolás M., Shifa Jebari-Benslaiman, Roberto Jiménez-Monzón, Asier Benito-Vicente, Yeray Brito-Casillas, Laida Garcés, Ana M. González-Lleo, Antonio Tugores, Mauro Boronat, César Martin, and et al. 2023. "Age, Origin and Functional Study of the Prevalent LDLR Mutation Causing Familial Hypercholesterolaemia in Gran Canaria" International Journal of Molecular Sciences 24, no. 14: 11319. https://doi.org/10.3390/ijms241411319
APA StyleSuárez, N. M., Jebari-Benslaiman, S., Jiménez-Monzón, R., Benito-Vicente, A., Brito-Casillas, Y., Garcés, L., González-Lleo, A. M., Tugores, A., Boronat, M., Martin, C., Wägner, A. M., & Sánchez-Hernández, R. M. (2023). Age, Origin and Functional Study of the Prevalent LDLR Mutation Causing Familial Hypercholesterolaemia in Gran Canaria. International Journal of Molecular Sciences, 24(14), 11319. https://doi.org/10.3390/ijms241411319