
Hyperuricaemia Does Not Interfere with Aortopathy in a Murine Model of Marfan Syndrome

 , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
2. Results
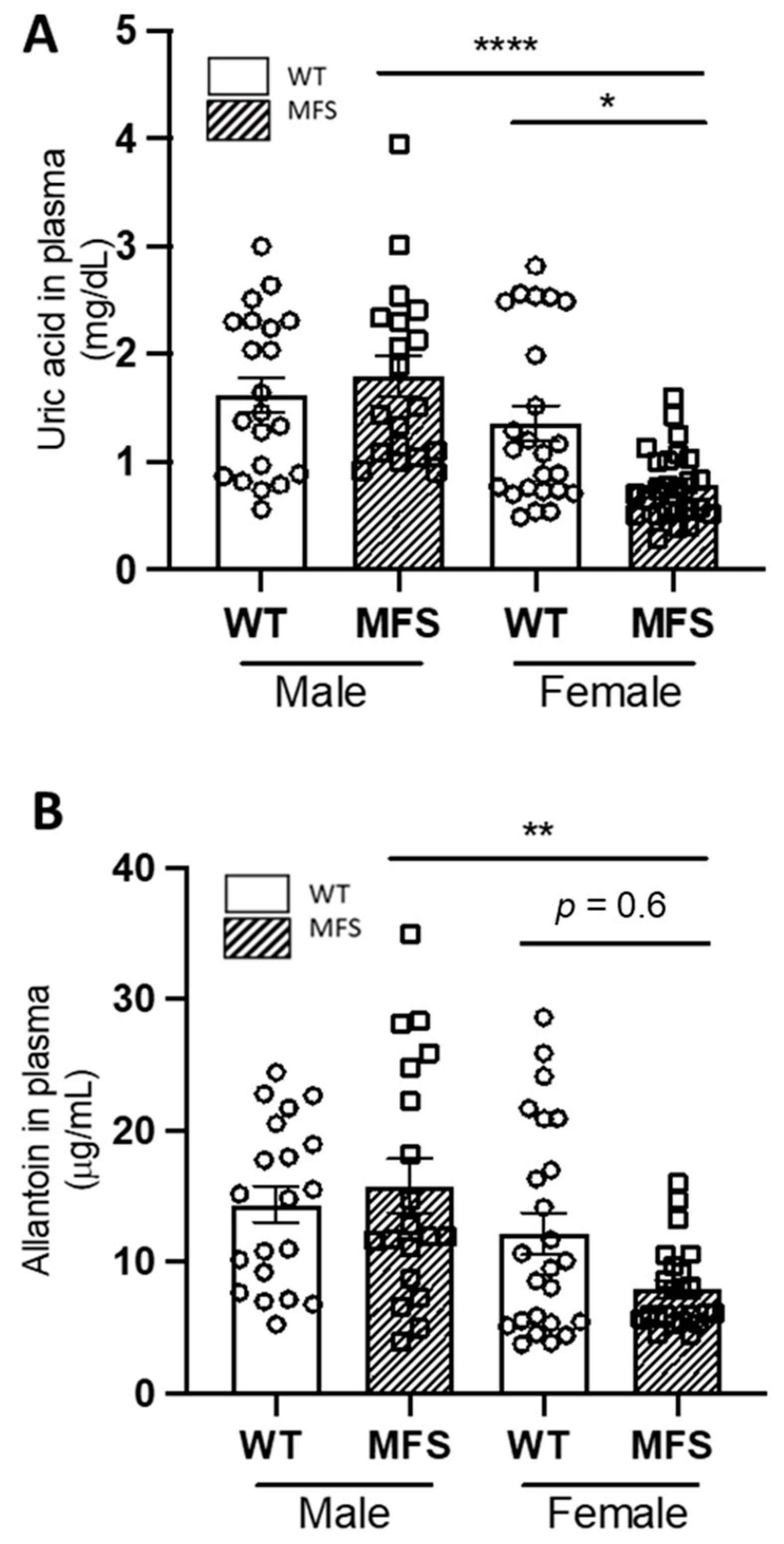
2.1. Sex Differences in Uric Acid and Allantoin Blood Plasma Levels in MFS Mice
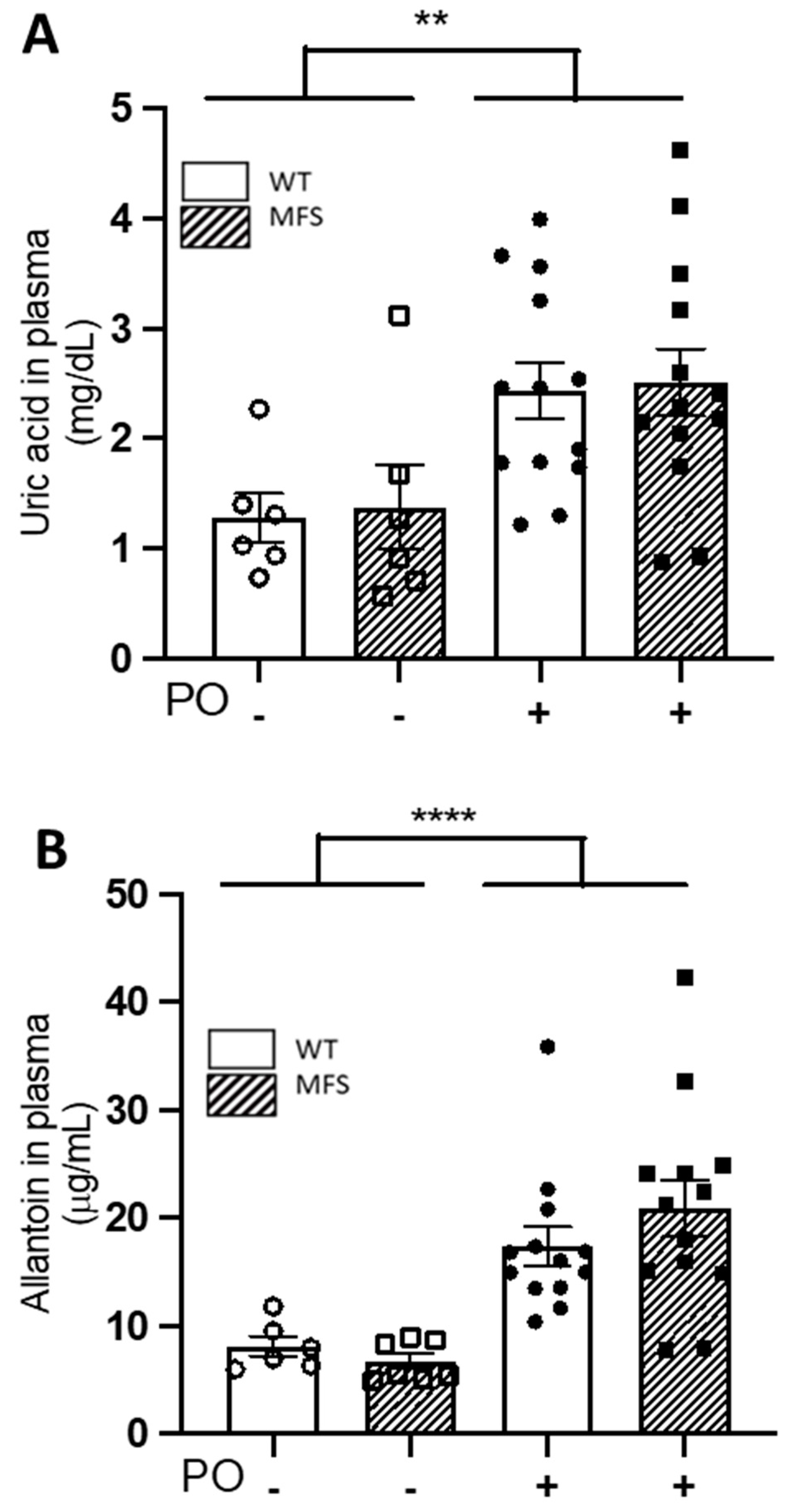
2.2. Potassium Oxonate Transiently Increased Blood Plasma Levels of Uric Acid Both in WT and MFS Mice
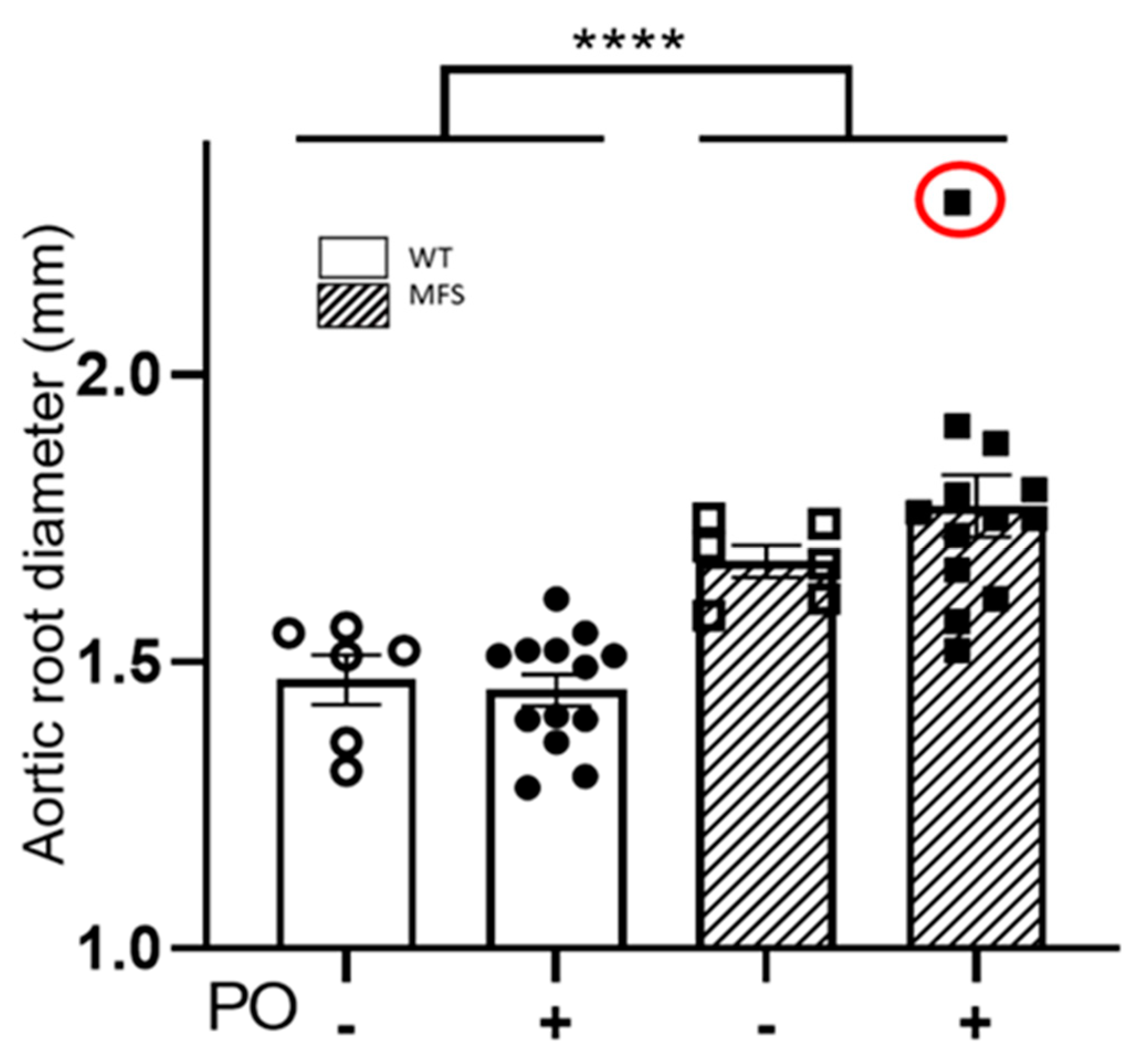
2.3. Aortopathy in MFS Mice Progressed Regardless of the Potassium Oxonate-Induced Hyperuricaemia
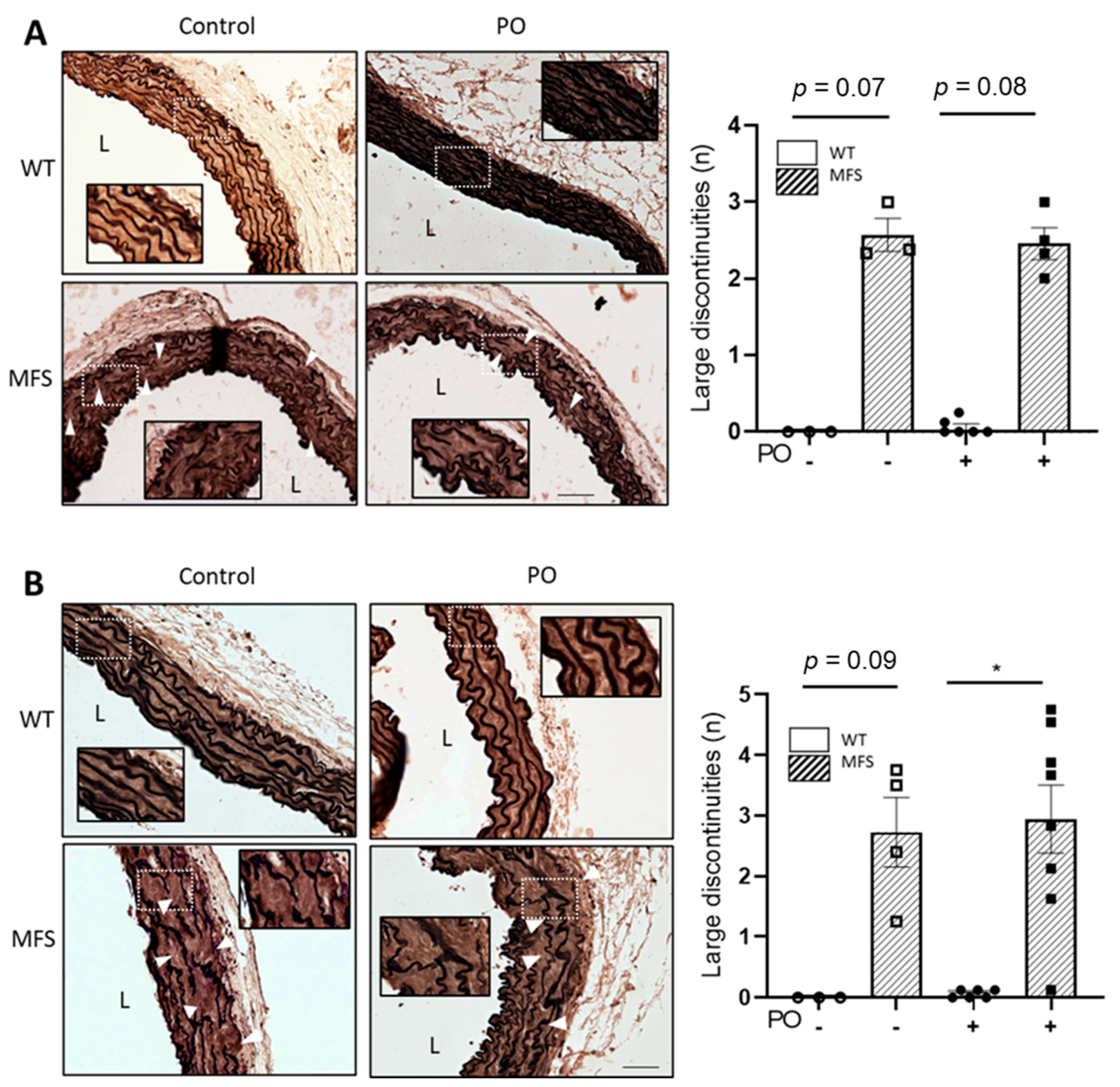
2.4. The Characteristic Structural Disarray of MFS Aorta Persisted following Potassium Oxonate-Induced Hyperuricaemia
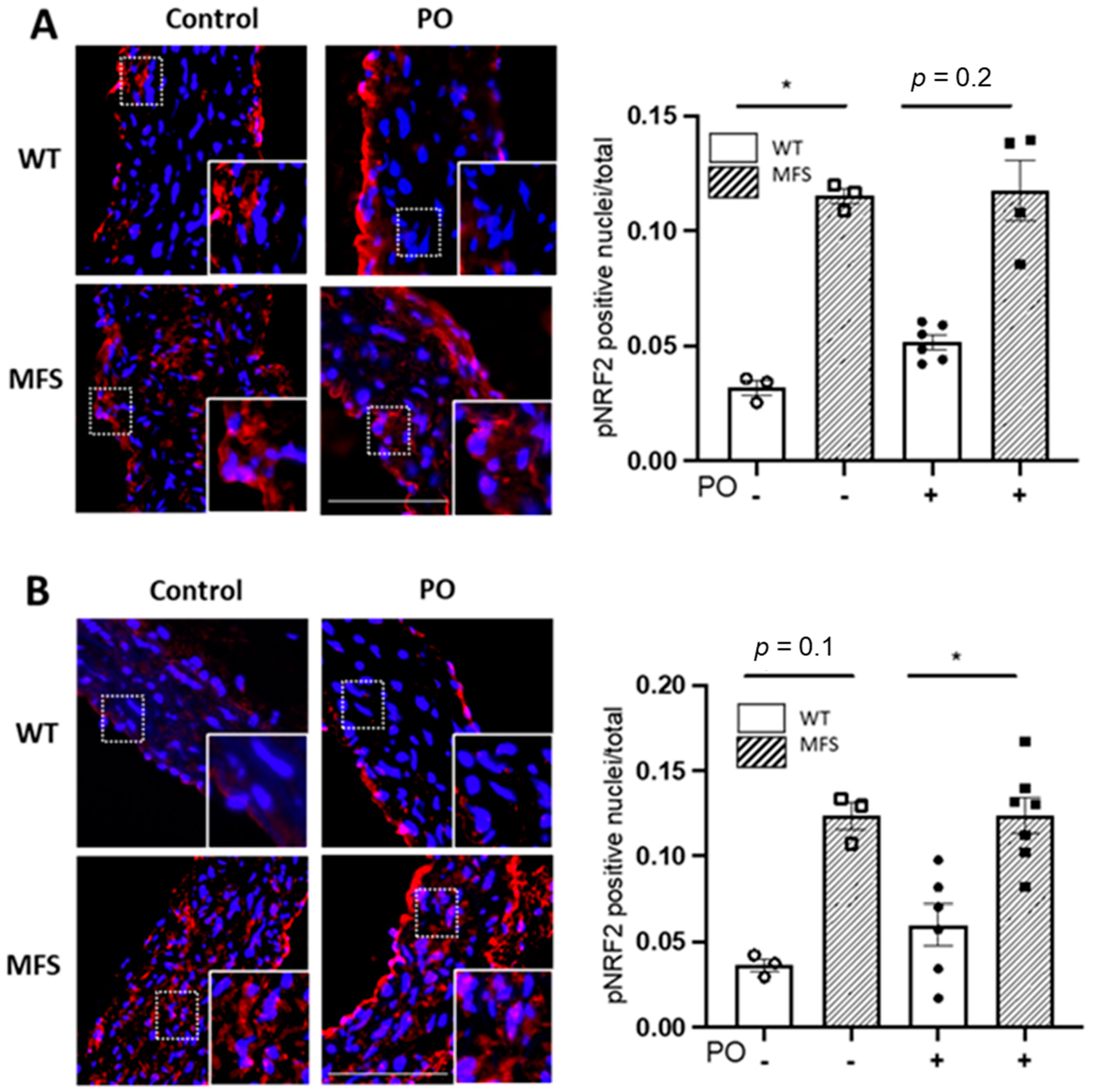
2.5. Potassium Oxonate-Induced Hyperuricaemia Does Not Modify Redox Stress Markers Changes Occurring in the MFS Aortic Media
2.6. Blood Pressure Is Not Perturbed by Potassium Oxonate Administration
3. Discussion
Limitations
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Mice, Experimental Design, and Study Approval
4.3. Uric Acid and Allantoin Analysis in Blood Plasma
4.4. Echocardiography
4.5. Histopathology
4.6. Immunofluorescence Staining
4.7. Blood Pressure Measurements
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef] [PubMed]
- Portelli, S.S.; Hambly, B.D.; Jeremy, R.W.; Robertson, E.N. Oxidative stress in genetically triggered thoracic aortic aneurysm: Role in pathogenesis and therapeutic opportunities. Redox Rep. 2021, 26, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Salmon, M. NADPH Oxidases in Aortic Aneurysms. Antioxidants 2022, 11, 1830. [Google Scholar] [CrossRef]
- Rysz, J.; Gluba-Brzózka, A.; Rokicki, R.; Franczyk, B. Oxidative Stress-Related Susceptibility to Aneurysm in Marfan’s Syndrome. Biomedicines 2021, 9, 1171. [Google Scholar] [CrossRef]
- Malecki, C.; Hambly, B.D.; Jeremy, R.W.; Robertson, E.N. The Role of Inflammation and Myeloperoxidase-Related Oxidative Stress in the Pathogenesis of Genetically Triggered Thoracic Aortic Aneurysms. Int. J. Mol. Sci. 2020, 21, 7678. [Google Scholar] [CrossRef]
- Pincemail, J.; Defraigne, J.O.; Courtois, A.; Albert, A.; Cheramy-Bien, J.P.; Sakalihasan, N. Abdominal Aortic Aneurysm (AAA): Is There a Role for the Prevention and Therapy Using Antioxidants? Curr. Drug Targets 2018, 19, 1256–1264. [Google Scholar] [CrossRef]
- Sánchez-Infantes, D.; Nus, M.; Navas-Madroñal, M.; Fité, J.; Pérez, B.; Barros-Membrilla, A.J.; Soto, B.; Martínez-González, J.; Camacho, M.; Rodriguez, C.; et al. Oxidative Stress and Inflammatory Markers in Abdominal Aortic Aneurysm. Antioxidants 2021, 10, 602. [Google Scholar] [CrossRef]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar] [CrossRef]
- Asano, K.; Cantalupo, A.; Sedes, L.; Ramirez, F. Pathophysiology and Therapeutics of Thoracic Aortic Aneurysm in Marfan Syndrome. Biomolecules 2022, 12, 128. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Braverman, A.C.; De Backer, J.; Morris, S.A.; Boileau, C.; Maumenee, I.H.; Jondeau, G.; Evangelista, A.; Pyeritz, R.E. Marfan syndrome. Nat. Rev. Dis. Primers 2021, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Demolder, A.; von Kodolitsch, Y.; Muiño-Mosquera, L.; De Backer, J. Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics 2020, 10, 751. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef]
- von Kodolitsch, Y.; Demolder, A.; Girdauskas, E.; Kaemmerer, H.; Kornhuber, K.; Muino Mosquera, L.; Morris, S.; Neptune, E.; Pyeritz, R.; Rand-Hendriksen, S.; et al. Features of Marfan syndrome not listed in the Ghent nosology—The dark side of the disease. Expert Rev. Cardiovasc. Ther. 2019, 17, 883–915. [Google Scholar] [CrossRef]
- Soto, M.E.; Ochoa-Hein, E.; Anaya-Ayala, J.E.; Ayala-Picazo, M.; Koretzky, S.G. Systematic review and meta-analysis of aortic valve-sparing surgery versus replacement surgery in ascending aortic aneurysms and dissection in patients with Marfan syndrome and other genetic connective tissue disorders. J. Thorac. Dis. 2021, 13, 4830–4844. [Google Scholar] [CrossRef]
- Steinmetz, L.M.; Coselli, J.S. Endovascular Repair in Patients with Marfan Syndrome: Concerns Amid Controversy. Ann. Vasc. Surg. 2022, 17, S0890-5096(22)00236-9. [Google Scholar] [CrossRef]
- Moshirfar, M.; Barke, M.R.; Huynh, R.; Waite, A.J.; Ply, B.; Ronquillo, Y.C.; Hoopes, P.C. Controversy and Consideration of Refractive Surgery in Patients with Heritable Disorders of Connective Tissue. J. Clin. Med. 2021, 10, 3769. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, A.; Spata, E.; Emberson, J.; Davies, K.; Halls, H.; Holland, L.; Wilson, K.; Reith, C.; Child, A.H.; Clayton, T.; et al. Angiotensin receptor blockers and β blockers in Marfan syndrome: An individual patient data meta-analysis of randomised trials. Lancet 2022, 400, 822–831. [Google Scholar] [CrossRef]
- Hibender, S.; Franken, R.; van Roomen, C.; Ter Braake, A.; van der Made, I.; Schermer, E.E.; Gunst, Q.; van den Hoff, M.J.; Lutgens, E.; Pinto, Y.M.; et al. Resveratrol Inhibits Aortic Root Dilatation in the Fbn1C1039G/+ Marfan Mouse Model. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1618–1626. [Google Scholar] [CrossRef]
- van Andel, M.M.; Groenink, M.; Zwinderman, A.H.; Mulder, B.J.M.; de Waard, V. The Potential Beneficial Effects of Resveratrol on Cardiovascular Complications in Marfan Syndrome Patients-Insights from Rodent-Based Animal Studies. Int. J. Mol. Sci. 2019, 20, 1122. [Google Scholar] [CrossRef]
- Mieremet, A.; van der Stoel, M.; Li, S.; Coskun, E.; van Krimpen, T.; Huveneers, S.; de Waard, V. Endothelial dysfunction in Marfan syndrome mice is restored by resveratrol. Sci. Rep. 2022, 12, 22504. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rovira, I.; Arce, C.; De Rycke, K.; Pérez, B.; Carretero, A.; Arbonés, M.; Teixidò-Turà, G.; Gómez-Cabrera, M.C.; Campuzano, V.; Jiménez-Altayó, F.; et al. Allopurinol blocks aortic aneurysm in a mouse model of Marfan syndrome via reducing aortic oxidative stress. Free Radic. Biol. Med. 2022, 193, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, H.; Luo, C.; Zhao, Y.; Dai, R.; Li, Z.; Zhang, X.; Gong, Z.; Cai, Z.; Shen, Y.; et al. Urate-Lowering Therapy Inhibits Thoracic Aortic Aneurysm and Dissection Formation in Mice. Arterioscler. Thromb. Vasc. Biol. 2023, in press. [Google Scholar] [CrossRef]
- Schwaerzer, G.K.; Kalyanaraman, H.; Casteel, D.E.; Dalton, N.D.; Gu, Y.; Lee, S.; Zhuang, S.; Wahwah, N.; Schilling, J.M.; Patel, H.H.; et al. Aortic pathology from protein kinase G activation is prevented by an antioxidant vitamin B12 analog. Nat. Commun. 2019, 10, 3533. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Altayó, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Del Blanco, D.G.; López-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barberà, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Y.; Siu, K.L.; Zhang, Y.; Cai, H. Targeting feed-forward signaling of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis with anti-TGFβ and folic acid attenuates formation of aortic aneurysms: Novel mechanisms and therapeutics. Redox Biol. 2021, 38, 101757. [Google Scholar] [CrossRef]
- Yu, W.; Xiao, L.; Que, Y.; Li, S.; Chen, L.; Hu, P.; Xiong, R.; Seta, F.; Chen, H.; Tong, X. Smooth muscle NADPH oxidase 4 promotes angiotensin II-induced aortic aneurysm and atherosclerosis by regulating osteopontin. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165912. [Google Scholar] [CrossRef]
- Toral, M.; de la Fuente-Alonso, A.; Campanero, M.R.; Redondo, J.M. The NO signalling pathway in aortic aneurysm and dissection. Br. J. Pharmacol. 2022, 179, 1287–1303. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Ruiz-Rodríguez, M.J.; Desdín-Micó, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuña, P.; et al. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Siegert, A.M.; García Díaz-Barriga, G.; Esteve-Codina, A.; Navas-Madroñal, M.; Gorbenko Del Blanco, D.; Alberch, J.; Heath, S.; Galán, M.; Egea, G. A FBN1 3′UTR mutation variant is associated with endoplasmic reticulum stress in aortic aneurysm in Marfan syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 107–114. [Google Scholar] [CrossRef]
- Meirelles, T.; Araujo, T.L.S.; Nolasco, P.; Moretti, A.I.S.; Guido, M.C.; Debbas, V.; Pereira, L.V.; Laurindo, F.R. Fibrillin-1 mgΔ(lpn) Marfan syndrome mutation associates with preserved proteostasis and bypass of a protein disulfide isomerase-dependent quality checkpoint. Int. J. Biochem. Cell Biol. 2016, 71, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: A leading actor in cardiovascular disease drama. Redox Biol. 2021, 48, 102195. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.H.; van Breemen, C.; Chung, A.W. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vasc. Pharmacol. 2010, 52, 37–45. [Google Scholar] [CrossRef]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Seet, R.C.; Kasiman, K.; Gruber, J.; Tang, S.Y.; Wong, M.C.; Chang, H.M.; Chan, Y.H.; Halliwell, B.; Chen, C.P. Is uric acid protective or deleterious in acute ischemic stroke? A prospective cohort study. Atherosclerosis 2010, 209, 215–219. [Google Scholar] [CrossRef]
- Kanbay, M.; Segal, M.; Afsar, B.; Kang, D.H.; Rodriguez-Iturbe, B.; Johnson, R.J. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart 2013, 99, 759–766. [Google Scholar] [CrossRef]
- Vassalle, C.; Mazzone, A.; Sabatino, L.; Carpeggiani, C. Uric Acid for Cardiovascular Risk: Dr. Jekyll or Mr. Hide? Diseases 2016, 4, 12. [Google Scholar] [CrossRef]
- Masi, S.; Pugliese, N.R.; Taddei, S. The difficult relationship between uric acid and cardiovascular disease. Eur. Heart J. 2019, 40, 3055–3057. [Google Scholar] [CrossRef]
- Yu, W.; Cheng, J.D. Uric Acid and Cardiovascular Disease: An Update from Molecular Mechanism to Clinical Perspective. Front. Pharmacol. 2020, 11, 582680. [Google Scholar] [CrossRef]
- Lee, S.J.; Oh, B.K.; Sung, K.C. Uric acid and cardiometabolic diseases. Clin. Hypertens. 2020, 26, 13. [Google Scholar] [CrossRef] [PubMed]
- Cortese, F.; Giordano, P.; Scicchitano, P.; Faienza, M.F.; De Pergola, G.; Calculli, G.; Meliota, G.; Ciccone, M.M. Uric acid: From a biological advantage to a potential danger. A focus on cardiovascular effects. Vasc. Pharmacol. 2019, 120, 106565. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Ha, S.K. Uric Acid Puzzle: Dual Role as Anti-oxidant and Pro-oxidant. Electrolytes Blood Press. 2014, 12, 1–6. [Google Scholar] [CrossRef]
- Glantzounis, G.K.; Tsimoyiannis, E.C.; Kappas, A.M.; Galaris, D.A. Uric acid and oxidative stress. Curr. Pharm. Des. 2005, 11, 4145–4151. [Google Scholar] [CrossRef]
- Itahana, Y.; Han, R.; Barbier, S.; Lei, Z.; Rozen, S.; Itahana, K. The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene 2015, 34, 1799–1810. [Google Scholar] [CrossRef]
- Scott, G.S.; Hooper, D.C. The role of uric acid in protection against peroxynitrite-mediated pathology. Med. Hypotheses 2001, 56, 95–100. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef] [PubMed]
- Corry, D.B.; Eslami, P.; Yamamoto, K.; Nyby, M.D.; Makino, H.; Tuck, M.L. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J. Hypertens. 2008, 26, 269–275. [Google Scholar] [CrossRef]
- Yu, M.A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Patetsios, P.; Rodino, W.; Wisselink, W.; Bryan, D.; Kirwin, J.D.; Panetta, T.F. Identification of uric acid in aortic aneurysms and atherosclerotic artery. Ann. N. Y. Acad. Sci. 1996, 800, 243–245. [Google Scholar] [CrossRef]
- Esen, A.M.; Akcakoyun, M.; Esen, O.; Acar, G.; Emiroglu, Y.; Pala, S.; Kargin, R.; Karapinar, H.; Ozcan, O.; Barutcu, I. Uric acid as a marker of oxidative stress in dilatation of the ascending aorta. Am. J. Hypertens. 2011, 24, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, Y.; Zou, J.; Shen, Y.; Luo, D.; Bao, H.; Chen, Y.; Ye, J.; Guan, J.L. Serum uric acid could be served as an independent marker for increased risk and severity of ascending aortic dilatation in Behçet’s disease patients. J. Clin. Lab. Anal. 2019, 33, e22637. [Google Scholar] [CrossRef] [PubMed]
- Otaki, Y.; Watanabe, T.; Konta, T.; Watanabe, M.; Asahi, K.; Yamagata, K.; Fujimoto, S.; Tsuruya, K.; Narita, I.; Kasahara, M.; et al. Impact of hyperuricemia on mortality related to aortic diseases: A 3.8-year nationwide community-based cohort study. Sci. Rep. 2020, 10, 14281. [Google Scholar] [CrossRef]
- Yang, G.; Chai, X.; Ding, N.; Yang, D.; Ding, Q. A retrospective observational study of serum uric acid and in-hospital mortality in acute type A aortic dissection. Sci. Rep. 2022, 12, 12289. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, A.; Kuo, A.; Frishman, W.H. Allopurinol as a cardiovascular drug. Cardiol. Rev. 2011, 19, 265–271. [Google Scholar] [CrossRef]
- Høieggen, A.; Alderman, M.H.; Kjeldsen, S.E.; Julius, S.; Devereux, R.B.; De Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristianson, K.; Lederballe-Pedersen, O.; et al. The impact of serum uric acid on cardiovascular outcomes in the LIFE study. Kidney Int. 2004, 65, 1041–1049. [Google Scholar] [CrossRef]
- Hamada, T.; Ichida, K.; Hosoyamada, M.; Mizuta, E.; Yanagihara, K.; Sonoyama, K.; Sugihara, S.; Igawa, O.; Hosoya, T.; Ohtahara, A.; et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am. J. Hypertens. 2008, 21, 1157–1162. [Google Scholar] [CrossRef]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef]
- Delbosc, S.; Paizanis, E.; Magous, R.; Araiz, C.; Dimo, T.; Cristol, J.P.; Cros, G.; Azay, J. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 2005, 179, 43–49. [Google Scholar] [CrossRef]
- Nyby, M.D.; Abedi, K.; Smutko, V.; Eslami, P.; Tuck, M.L. Vascular Angiotensin type 1 receptor expression is associated with vascular dysfunction, oxidative stress and inflammation in fructose-fed rats. Hypertens. Res. 2007, 30, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C. The role of uric acid in the development of cardiovascular disease. Curr. Med. Res. Opin. 2015, 31 (Suppl. S2), 1–2. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Song, Y.; Yan, Y.; Ding, Z. Elevated serum uric acid and risk of cardiovascular or all-cause mortality in people with suspected or definite coronary artery disease: A meta-analysis. Atherosclerosis 2016, 254, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, Y.; Ogino, K.; Furuse, Y.; Shiomi, T.; Tsutsui, H.; Yamamoto, T.; Igawa, O.; Hisatome, I.; Shigemasa, C. Allopurinol improves cardiac dysfunction after ischemia-reperfusion via reduction of oxidative stress in isolated perfused rat hearts. Circ. J. 2003, 67, 781–787. [Google Scholar] [CrossRef]
- Hooper, D.C.; Kean, R.B.; Scott, G.S.; Spitsin, S.V.; Mikheeva, T.; Morimoto, K.; Bette, M.; Röhrenbeck, A.M.; Dietzschold, B.; Weihe, E. The central nervous system inflammatory response to neurotropic virus infection is peroxynitrite dependent. J. Immunol. 2001, 167, 3470–3477. [Google Scholar] [CrossRef]
- Lu, J.; Dalbeth, N.; Yin, H.; Li, C.; Merriman, T.R.; Wei, W.H. Mouse models for human hyperuricaemia: A critical review. Nat. Rev. Rheumatol. 2019, 15, 413–426. [Google Scholar] [CrossRef]
- Lin, C.; Zheng, Q.; Li, Y.; Wu, T.; Luo, J.; Jiang, Y.; Huang, Q.; Yan, C.; Zhang, L.; Zhang, W.; et al. Assessment of the influence on left ventricle by potassium oxonate and hypoxanthine-induced chronic hyperuricemia. Exp. Biol. Med. 2023, 248, 165–174. [Google Scholar] [CrossRef]
- Qian, X.; Wang, X.; Luo, J.; Liu, Y.; Pang, J.; Zhang, H.; Xu, Z.; Xie, J.; Jiang, X.; Ling, W. Hypouricemic and nephroprotective roles of anthocyanins in hyperuricemic mice. Food Funct. 2019, 10, 867–878. [Google Scholar] [CrossRef]
- Yasutake, Y.; Tomita, K.; Higashiyama, M.; Furuhashi, H.; Shirakabe, K.; Takajo, T.; Maruta, K.; Sato, H.; Narimatsu, K.; Yoshikawa, K.; et al. Uric acid ameliorates indomethacin-induced enteropathy in mice through its antioxidant activity. J. Gastroenterol. Hepatol. 2017, 32, 1839–1845. [Google Scholar] [CrossRef]
- Cutler, R.G.; Camandola, S.; Feldman, N.H.; Yoon, J.S.; Haran, J.B.; Arguelles, S.; Mattson, M.P. Uric acid enhances longevity and endurance and protects the brain against ischemia. Neurobiol. Aging 2019, 75, 159–168. [Google Scholar] [CrossRef]
- Cutler, R.G.; Camandola, S.; Malott, K.F.; Edelhauser, M.A.; Mattson, M.P. The Role of Uric Acid and Methyl Derivatives in the Prevention of Age-Related Neurodegenerative Disorders. Curr. Top. Med. Chem. 2015, 15, 2233–2238. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Chung Wy Tomlinson, B. Simultaneous measurement of allantoin and urate in plasma: Analytical evaluation and potential clinical application in oxidant:antioxidant balance studies. Clin. Chem. 1999, 45, 901–904. [Google Scholar] [CrossRef]
- Wang, J.C.; Tsai, S.H.; Tsai, H.Y.; Lin, S.J.; Huang, P.H. Hyperuricemia exacerbates abdominal aortic aneurysm formation through the URAT1/ERK/MMP-9 signaling pathway. BMC Cardiovasc. Disord. 2023, 23, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, X.; Lu, Y.; Guo, L.; Ma, L. Preoperative uric acid predicts in-hospital death in patients with acute type A aortic dissection. J. Cardiothorac. Surg. 2020, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.L.; Qi, X.; Li, X.; Zhang, Y.F.; Xia, Q.Q.; Chen, J.C. Serum uric acid is associated with aortic dissection in Chinese men. Int. J. Cardiol. 2016, 202, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, H.; Yan, F.; He, Y.; Ji, A.; Liu, Z.; Li, M.; Ji, X.; Li, C. Kidney and plasma metabolomics provide insights into the molecular mechanisms of urate nephropathy in a mouse model of hyperuricemia. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166374. [Google Scholar] [CrossRef]
- Gherghina, M.E.; Peride, I.; Tiglis, M.; Neagu, T.P.; Niculae, A.; Checherita, I.A. Uric Acid and Oxidative Stress-Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int. J. Mol. Sci. 2022, 23, 3188. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Meirelles, T.; López-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko Del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizábal, Y.; et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef]
- Pedroza, A.J.; Tashima, Y.; Shad, R.; Cheng, P.; Wirka, R.; Churovich, S.; Nakamura, K.; Yokoyama, N.; Cui, J.Z.; Iosef, C.; et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2195–2211. [Google Scholar] [CrossRef]
- Onetti, Y.; Jiménez-Altayó, F.; Heras, M.; Vila, E.; Dantas, A.P. Western-type diet induces senescence, modifies vascular function in non-senescence mice and triggers adaptive mechanisms in senescent ones. Exp. Gerontol. 2013, 48, 1410–1419. [Google Scholar] [CrossRef]
- Arce, C.; Rodríguez-Rovira, I.; De Rycke, K.; Durán, K.; Campuzano, V.; Fabregat, I.; Jiménez-Altayó, F.; Berraondo, P.; Egea, G. Anti-TGFβ (Transforming Growth Factor β) Therapy with Betaglycan-Derived P144 Peptide Gene Delivery Prevents the Formation of Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e440–e452. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | WT | WT | WT | MFS | MFS | MFS | MFS | ||
|---|---|---|---|---|---|---|---|---|---|
| Potassium Oxanate (PO) | Mice Age (Months)/PO (Weeks) | IVSD | LVPWD | LVDD | LVDS | IVSD | LVPWD | LVDD | LVDS |
| ------ | 2 m/0 w | 0.82 ± 0.01 | 0.79 ± 0.01 | 1.98 ± 0.02 | 1.27 ± 0.03 | 0.81 ± 0.01 | 0.77 ± 0.01 | 1.98 ± 0.03 | 1.25 ± 0.03 |
| - | 3 m/4 w | 0.80 ± 0.02 | 0.76 ± 0.03 | 2.07 ± 0.11 | 1.27 ± 0.04 | 0.80 ± 0.02 | 0.78 ± 0.02 | 2.06 ± 0.04 | 1.36 ± 0.04 |
| - | 4 m/8 w | 0.87 ± 0.04 | 0.95 ± 0.09 | 1.88 ± 0.05 | 1.29 ± 0.08 | 0.95 ± 0.09 | 0.95 ± 0.09 | 2.31 ± 0.19 | 1.39 ± 0.05 |
| - | 6 m/16 w | 0.87 ± 0.03 | 0.85 ± 0.03 | 2.05 ± 0.07 | 1.40 ± 0.04 | 0.87 ± 0.01 | 0.83 ± 0.02 | 2.01 ± 0.03 | 1.32 ± 0.01 |
| + | 3 m/4 w | 0.83 ± 0.01 | 0.80 ± 0.002 | 2.03 ± 0.05 | 1.38 ± 0.02 | 0.82 ± 0.01 | 0.81 ± 0.01 | 2.07 ± 0.06 | 1.41 ± 0.04 |
| + | 4 m/8 w | 0.84 ± 0.04 | 0.93 ± 0.02 | 2.21 ± 0.19 | 1.43 ± 0.07 | 1.01 ± 0.05 | 1.06 ± 0.02 | 2.52 ± 0.07 | 1.71 ± 0.08 |
| + | 6 m/16 w | 0.79 ± 0.01 | 0.80 ± 0.02 | 2.11 ± 0.06 | 1.35 ± 0.04 | 0.79 ± 0.02 | 0.75 ± 0.03 | 2.00 ± 0.06 | 1.34 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Rovira, I.; López-Sainz, A.; Palomo-Buitrago, M.E.; Pérez, B.; Jiménez-Altayó, F.; Campuzano, V.; Egea, G. Hyperuricaemia Does Not Interfere with Aortopathy in a Murine Model of Marfan Syndrome. Int. J. Mol. Sci. 2023, 24, 11293. https://doi.org/10.3390/ijms241411293
Rodríguez-Rovira I, López-Sainz A, Palomo-Buitrago ME, Pérez B, Jiménez-Altayó F, Campuzano V, Egea G. Hyperuricaemia Does Not Interfere with Aortopathy in a Murine Model of Marfan Syndrome. International Journal of Molecular Sciences. 2023; 24(14):11293. https://doi.org/10.3390/ijms241411293
Chicago/Turabian StyleRodríguez-Rovira, Isaac, Angela López-Sainz, Maria Encarnación Palomo-Buitrago, Belen Pérez, Francesc Jiménez-Altayó, Victoria Campuzano, and Gustavo Egea. 2023. "Hyperuricaemia Does Not Interfere with Aortopathy in a Murine Model of Marfan Syndrome" International Journal of Molecular Sciences 24, no. 14: 11293. https://doi.org/10.3390/ijms241411293
APA StyleRodríguez-Rovira, I., López-Sainz, A., Palomo-Buitrago, M. E., Pérez, B., Jiménez-Altayó, F., Campuzano, V., & Egea, G. (2023). Hyperuricaemia Does Not Interfere with Aortopathy in a Murine Model of Marfan Syndrome. International Journal of Molecular Sciences, 24(14), 11293. https://doi.org/10.3390/ijms241411293