


Coronary “Microvascular Dysfunction”: Evolving Understanding of Pathophysiology, Clinical Implications, and Potential Therapeutics

 , , ,
, , , 
Abstract
1. Introduction
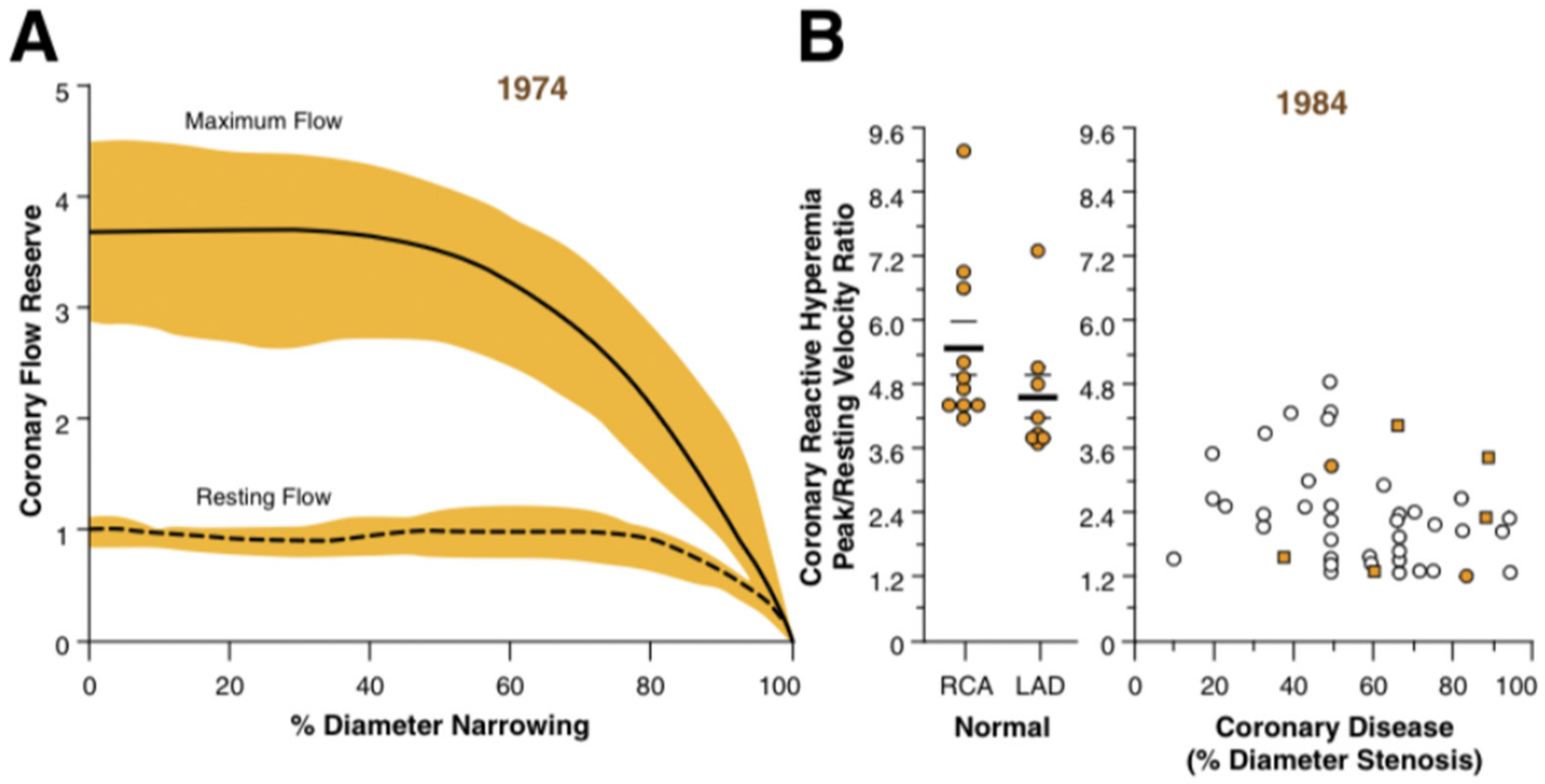
2. Insights from Periprocedural Physiological Studies
3. ORBITA and ISCHEMIA Studies: Efficacy of PCI
- The primary endpoint consisted of any of the following: cardiovascular death, AMI, unstable angina, heart failure and resuscitated cardiac arrest;
- Heart failure and unstable angina were only considered if they led to hospitalisation.
4. Assessment of Coronary Microcirculation
5. Coronary Microvascular Dysfunction: Can We Be More Precise?
- (a)
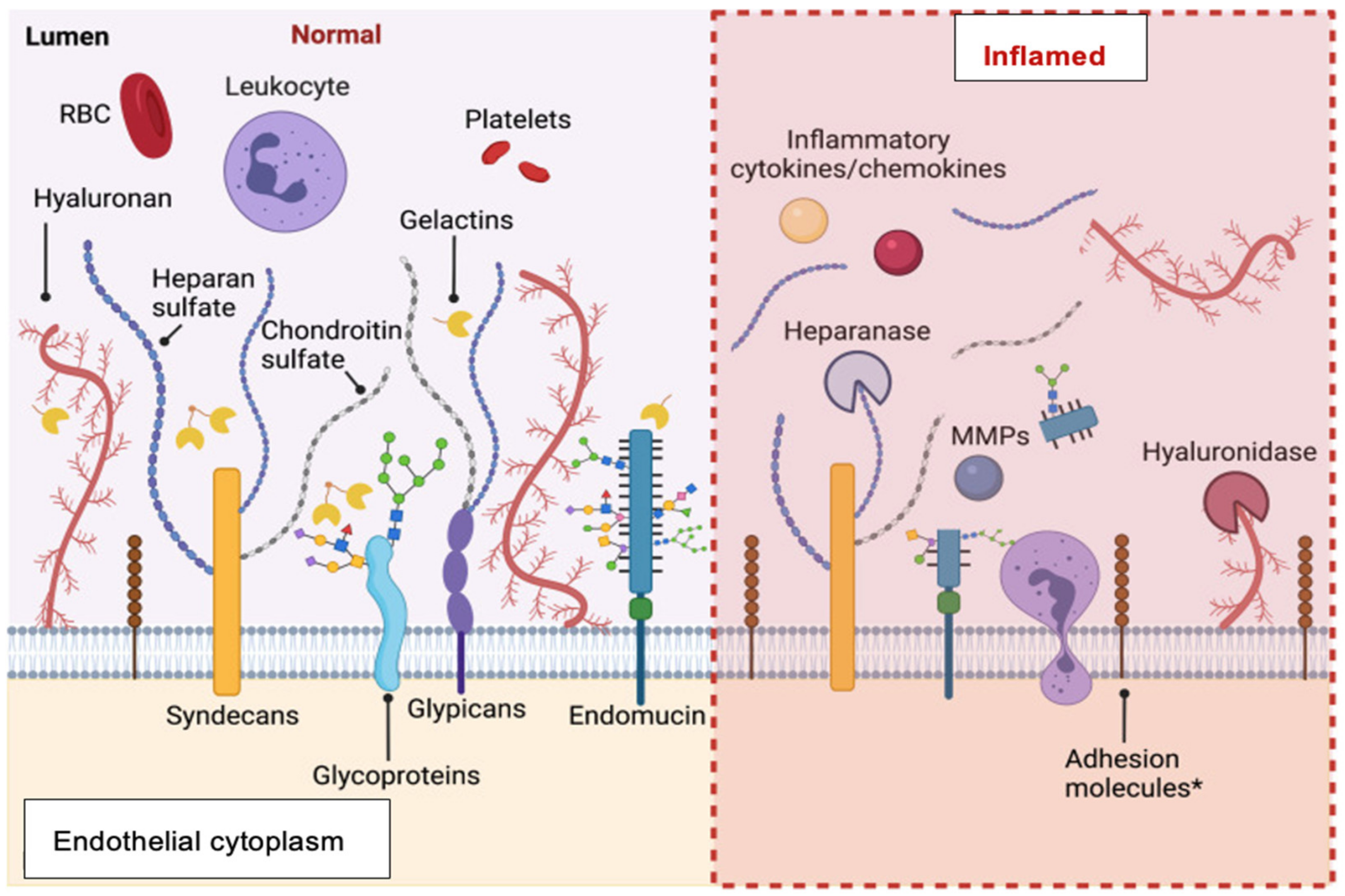
- Structural anomalies: Focus on the endothelial glycocalyx
- (b)
- Physiological anomalies: Focus on the endothelial dysfunction
- (c)
- Importance of vascular interactions with mast cell products and with circulating platelets
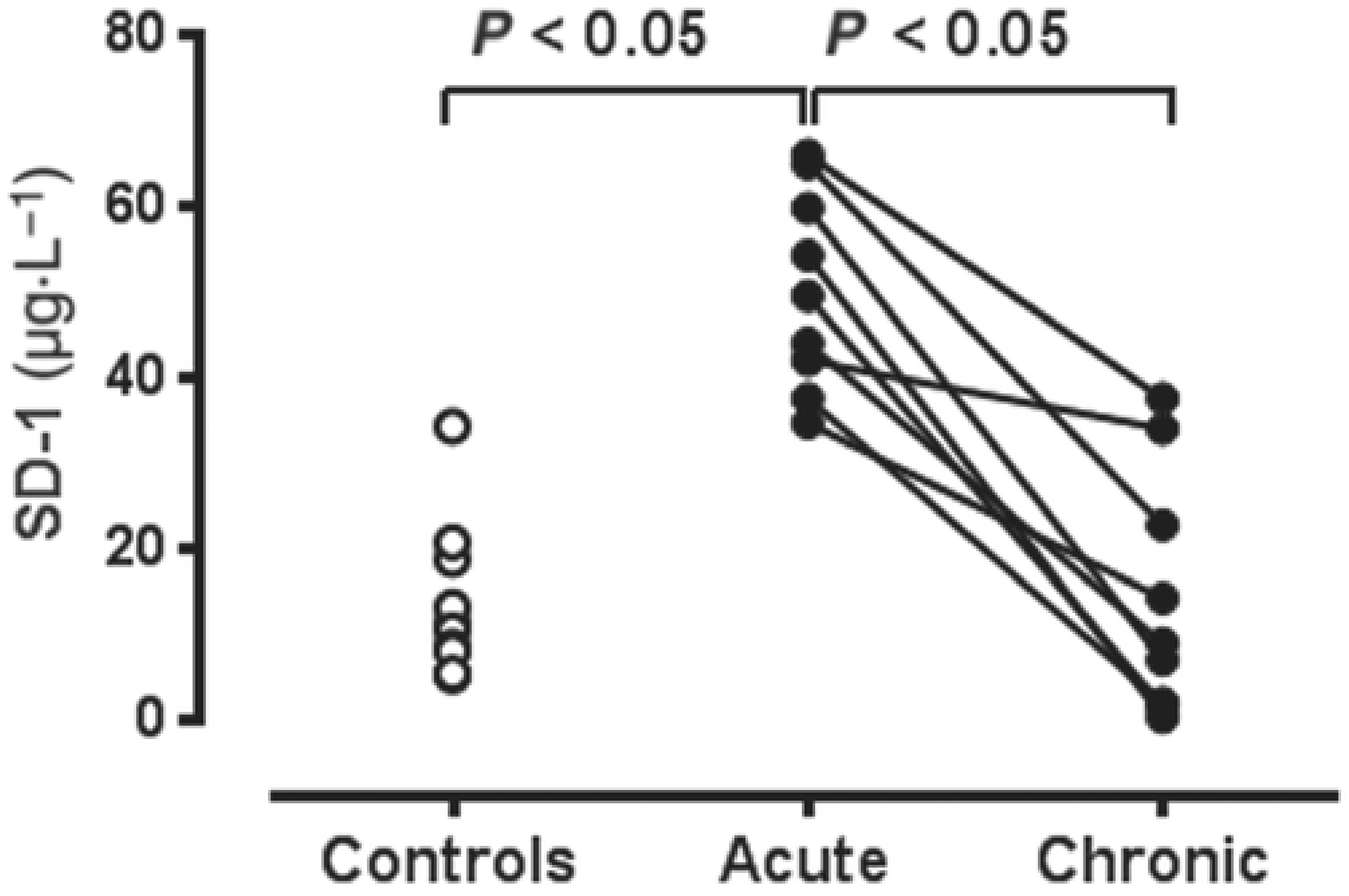
6. Some Examples of Pathophysiological Changes within the Coronary Microcirculation
- (a)
- Myocardial ischaemia
- (b)
- Normal Ageing
- (c)
- Diabetes Mellitus (DM)
- (d)
- Heart failure with preserved ejection fraction (HFpEF)
- (e)
- COVID-19-induced vasculitis.
7. Therapeutic Implications
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gould, K.; Lipscomb, K.; Hamilton, G.W. Physiologic basis for assessing critical coronary stenosis: Instantaneous flow response and regional distribution during coronary hyperemia as measures of coronary flow reserve. Am. J. Cardiol. 1974, 33, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Gould, K.L. Does coronary flow trump coronary anatomy? JACC Cardiovasc. Imaging 2009, 2, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.A.; Crea, F. Primary coronary microvascular dysfunction: Clinical presentation, pathophysiology, and management. Circulation 2010, 121, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Kogame, N.; Ono, M.; Kawashima, H.; Tomaniak, M.; Hara, H.; Leipsic, J.; Andreini, D.; Collet, C.; Patel, M.R.; Tu, S.; et al. The Impact of Coronary Physiology on Contemporary Clinical Decision Making. JACC Cardiovasc. Interv. 2020, 13, 1617–1638. [Google Scholar] [CrossRef]
- De Bruyne, B.; Pijls, N.H.; Kalesan, B.; Barbato, E.; Tonino, P.A.; Piroth, Z.; Jagic, N.; Mobius-Winckler, S.; Rioufol, G.; Witt, N.; et al. Fractional Flow Reserve–Guided PCI versus Medical Therapy in Stable Coronary Disease. N. Engl. J. Med. 2012, 367, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Tonino, P.A.L.; De Bruyne, B.; Pijls, N.H.J.; Siebert, U.; Ikeno, F.; van’t Veer, M.; Klauss, V.; Manoharan, G.; Engstrøm, T.; Oldroyd, K.G.; et al. Fractional flow reserve versus angiography for guiding percutaneous coronary intervention. N. Engl. J. Med. 2009, 360, 213–224. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Kasula, S.; Hacioglu, Y.; Ahmed, Z.; Uretsky, B.F.; Hakeem, A. Utilizing Post-Intervention Fractional Flow Reserve to Optimize Acute Results and the Relationship to Long-Term Outcomes. JACC Cardiovasc. Interv. 2016, 9, 1022–1031. [Google Scholar] [CrossRef]
- Howard, J.P.; Murthy, V.L. A Song of Pressure and Flow, or There and Back Again. JACC Cardiovasc. Interv. 2018, 11, 754–756. [Google Scholar] [CrossRef]
- Lee, J.M.; Jung, J.-H.; Hwang, D.; Park, J.; Fan, Y.; Na, S.-H.; Doh, J.-H.; Nam, C.-W.; Shin, E.-S.; Koo, B.-K. Coronary Flow Reserve and Microcirculatory Resistance in Patients with Intermediate Coronary Stenosis. J. Am. Coll. Cardiol. 2016, 67, 1158–1169. [Google Scholar] [CrossRef]
- Pijls, N.H.; van Schaardenburgh, P.; Manoharan, G.; Boersma, E.; Bech, J.-W.; Veer, M.V.; Bär, F.; Hoorntje, J.; Koolen, J.; Wijns, W.; et al. Percutaneous Coronary Intervention of Functionally Nonsignificant Stenosis: 5-Year Follow-Up of the DEFER Study. J. Am. Coll. Cardiol. 2007, 49, 2105–2111. [Google Scholar] [CrossRef]
- Zimmermann, F.M.; Ferrara, A.; Johnson, N.P.; Van Nunen, L.X.; Escaned, J.; Albertsson, P.; Erbel, R.; Legrand, V.; Gwon, H.-C.; Remkes, W.S.; et al. Deferral vs. performance of percutaneous coronary intervention of functionally non-significant coronary stenosis: 15-year follow-up of the DEFER trial. Eur. Heart J. 2015, 36, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- De Bruyne, B.; Fearon, W.F.; Pijls, N.H.; Barbato, E.; Tonino, P.; Piroth, Z.; Jagic, N.; Mobius-Winckler, S.; Rioufol, G.; Witt, N.; et al. Fractional Flow Reserve–Guided PCI for Stable Coronary Artery Disease. N. Engl. J. Med. 2014, 371, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Xaplanteris, P.; Fournier, S.; Pijls, N.H.; Fearon, W.F.; Barbato, E.; Tonino, P.A.; Engstrøm, T.; Kääb, S.; Dambrink, J.-H.; Rioufol, G.; et al. Five-Year Outcomes with PCI Guided by Fractional Flow Reserve. N. Engl. J. Med. 2018, 379, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E. Which is more enduring—FAME or COURAGE? N. Engl. J. Med. 2012, 367, 1059–1061. [Google Scholar] [CrossRef]
- Rade, J.J. FFR-guided PCI—FAME may not be so fleeting after all. N. Engl. J. Med. 2014, 371, 1251–1253. [Google Scholar] [CrossRef]
- Al-Lamee, R.; Thompson, D.; Dehbi, H.-M.; Sen, S.; Tang, K.; Davies, J.; Keeble, T.; Mielewczik, M.; Kaprielian, R.; Malik, I.S.; et al. Percutaneous coronary intervention in stable angina (ORBITA): A double-blind, randomised controlled trial. Lancet 2017, 391, 31–40. [Google Scholar] [CrossRef]
- Maron, D.J.; Hochman, J.S.; Reynolds, H.R.; Bangalore, S.; O’Brien, S.M.; Boden, W.E.; Chaitman, B.R.; Senior, R.; López-Sendón, J.; Alexander, K.P.; et al. Initial Invasive or Conservative Strategy for Stable Coronary Disease. N. Engl. J. Med. 2020, 382, 1395–1407. [Google Scholar] [CrossRef]
- Lopez-Sendon, J.L.; Cyr, D.D.; Mark, D.B.; Bangalore, S.; Huang, Z.; White, H.; Alexander, K.P.; Li, J.; Nair, R.G.; Demkow, M.; et al. Effects of initial invasive vs. initial conservative treatment strategies on recurrent and total cardiovascular events in the ISCHEMIA trial. Eur. Heart J. 2022, 43, 148–149. [Google Scholar] [CrossRef]
- Chaitman, B.R.; Alexander, K.P.; Cyr, D.D.; Berger, J.S.; Reynolds, H.R.; Bangalore, S.; Boden, W.E.; Lopes, R.D.; Demkow, M.; Perna, G.P.; et al. Myocardial Infarction in the ISCHEMIA Trial: Impact of Different Definitions on Incidence, Prognosis, and Treatment Comparisons. Circulation 2021, 143, 790–804. [Google Scholar] [CrossRef]
- Prasad, A.; Gersh, B.J.; Bertrand, M.E.; Lincoff, A.M.; Moses, J.W.; Ohman, E.M.; White, H.D.; Pocock, S.J.; McLaurin, B.T.; Cox, D.A.; et al. Prognostic Significance of Periprocedural Versus Spontaneously Occurring Myocardial Infarction After Percutaneous Coronary Intervention in Patients with Acute Coronary Syndromes: An Analysis From the ACUITY (Acute Catheterization and Urgent Intervention Triage Strategy) Trial. J. Am. Coll. Cardiol. 2009, 54, 477–486. [Google Scholar] [CrossRef]
- Al-Lamee, R.; Howard, J.P.; Shun-Shin, M.J.; Thompson, D.; Dehbi, H.-M.; Sen, S.; Nijjer, S.; Petraco, R.; Davies, J.; Keeble, T.; et al. Fractional Flow Reserve and Instantaneous Wave-Free Ratio as Predictors of the Placebo-Controlled Response to Percutaneous Coronary Intervention in Stable Single-Vessel Coronary Artery Disease. Circulation 2018, 138, 1780–1792. [Google Scholar] [CrossRef] [PubMed]
- Fearon, W.F.; Balsam, L.B.; Farouque, H.M.O.; Robbins, R.C.; Fitzgerald, P.J.; Yock, P.G.; Yeung, A.C. Novel Index for Invasively Assessing the Coronary Microcirculation. Circulation 2003, 107, 3129–3132. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.L.; Naya, M.; Foster, C.R.; Hainer, J.; Gaber, M.; Di Carli, G.; Blankstein, R.; Dorbala, S.; Sitek, A.; Pencina, M.J.; et al. Improved cardiac risk assessment with noninvasive measures of coronary flow reserve. Circulation 2011, 124, 2215–2224. [Google Scholar] [CrossRef]
- Indorkar, R.; Kwong, R.Y.; Romano, S.; White, B.E.; Chia, R.C.; Trybula, M.; Evans, K.; Shenoy, C.; Farzaneh-Far, A. Global Coronary Flow Reserve Measured During Stress Cardiac Magnetic Resonance Imaging Is an Independent Predictor of Adverse Cardiovascular Events. JACC Cardiovasc. Imaging 2018, 12, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.L.; Bateman, T.M.; Beanlands, R.S.; Berman, D.S.; Borges-Neto, S.; Chareonthaitawee, P.; Cerqueira, M.D.; Dekemp, R.A.; DePuey, E.G.; Dilsizian, V.; et al. Clinical Quantification of Myocardial Blood Flow Using PET: Joint Position Paper of the SNMMI Cardiovascular Council and the ASNC. J. Nucl. Cardiol. 2017, 25, 269–297. [Google Scholar] [CrossRef]
- Melikian, N.; Vercauteren, S.; Fearon, W.; Cuisset, T.; MacCarthy, P.; Davidavicius, G.; Aarnoudse, W.; Bartunek, J.; Vanderheyden, M.; Wyffels, E.; et al. Quantitative assessment of coronary microvascular function in patients with and without epicardial atherosclerosis. Eurointervention 2010, 5, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Long, M.; Hu, X.; Huang, Z.; Hu, C.; Gao, X.; Du, Z. Thermodilution-Derived Coronary Microvascular Resistance and Flow Reserve in Patients with Cardiac Syndrome X. Circ. Cardiovasc. Interv. 2014, 7, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Solberg, O.G.; Ragnarsson, A.; Kvarsnes, A.; Endresen, K.; Kongsgård, E.; Aakhus, S.; Gullestad, L.; Stavem, K.; Aaberge, L. Reference interval for the index of coronary microvascular resistance. Eurointervention 2014, 9, 1069–1075. [Google Scholar] [CrossRef]
- Fearon, W.F.; Kobayashi, Y. Invasive Assessment of the Coronary Microvasculature: The Index of Microcirculatory Resistance. Circ. Cardiovasc. Interv. 2017, 10, e005361. [Google Scholar] [CrossRef]
- De Maria, G.L.; Oxford Acute Myocardial Infarction (OxAMI) Study investigators; Scarsini, R.; Shanmuganathan, M.; Kotronias, R.; Terentes-Printzios, D.; Borlotti, A.; Langrish, J.P.; Lucking, A.J.; Choudhury, R.P.; et al. Angiography-derived index of microcirculatory resistance as a novel, pressure-wire-free tool to assess coronary microcirculation in ST elevation myocardial infarction. Int. J. Cardiovasc. Imaging 2020, 36, 1395–1406. [Google Scholar] [CrossRef]
- Dai, N.; Che, W.; Liu, L.; Zhang, W.; Yin, G.; Xu, B.; Xu, Y.; Duan, S.; Yu, H.; Li, C.; et al. Diagnostic Value of Angiography-Derived IMR for Coronary Microcirculation and Its Prognostic Implication After PCI. Front. Cardiovasc. Med. 2021, 8, 735743. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Dai, N.; Li, Y.; Kim, J.; Shin, D.; Lee, S.H.; Joh, H.S.; Kim, H.K.; Jeon, K.-H.; Ha, S.J.; et al. Functional Coronary Angiography–Derived Index of Microcirculatory Resistance in Patients With ST-Segment Elevation Myocardial Infarction. JACC Cardiovasc. Interv. 2021, 14, 1670–1684. [Google Scholar] [CrossRef]
- Gibson, C.M.; Cannon, C.P.; Daley, W.L.; Dodge, J.T.; Alexander, B.; Marble, S.J.; McCabe, C.H.; Raymond, L.; Fortin, T.; Poole, W.K.; et al. TIMI frame count: A quantitative method of assessing coronary artery flow. Circulation 1996, 93, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Jolly, S.S.; Hamid, T.; Sharma, D.; Chiha, J.; Chan, W.; Fuchs, F.; Bui, S.; Gao, P.; Kassam, S.; et al. Myocardial blush and microvascular reperfusion following manual thrombectomy during percutaneous coronary intervention for ST elevation myocardial infarction: Insights from the TOTAL trial. Eur. Heart J. 2016, 37, 1891–1898. [Google Scholar] [CrossRef]
- Van’t Hof, A.W.; Liem, A.; Suryapranata, H.; Hoorntje, J.C.; de Boer, M.J.; Zijlstra, F. Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction: Myocardial blush grade. Zwolle Myocardial Infarction Study Group. Circulation 1998, 97, 2302–2306. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.-C.; Crea, F.; Gersh, B.J.; Camici, P.G. Reappraisal of Ischemic Heart Disease. Circulation 2018, 138, 1463–1480. [Google Scholar] [CrossRef] [PubMed]
- Taqueti, V.R.; Di Carli, M.F. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2625–2641. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Montone, R.A.; Camilli, M.; Carbone, S.; Narula, J.; Lavie, C.J.; Niccoli, G.; Crea, F. Coronary Microvascular Dysfunction across the Spectrum of Cardiovascular Diseases: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1352–1371. [Google Scholar] [CrossRef]
- Kuebler, W.M.; Habazettl, H. Coronary Microcirculation in Ischemic Heart Disease. Curr. Pharm. Des. 2018, 24, 2893–2899. [Google Scholar] [CrossRef]
- Annecke, T.; Fischer, J.; Hartmann, H.; Tschoep, J.; Rehm, M.; Conzen, P.; Sommerhoff, C.; Becker, B. Shedding of the coronary endothelial glycocalyx: Effects of hypoxia/reoxygenation vs. ischaemia/reperfusion. Br. J. Anaesth. 2011, 107, 679–686. [Google Scholar] [CrossRef]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.J.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Lipowsky, H.H. Microvascular Rheology and Hemodynamics. Microcirculation 2005, 12, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, R.; Lu, X.; Kassab, G.S. Role of glycocalyx in flow-induced production of nitric oxide and reactive oxygen species. Free. Radic. Biol. Med. 2009, 47, 600–607. [Google Scholar] [CrossRef]
- Lambadiari, V.; Kousathana, F.; Raptis, A.; Katogiannis, K.; Kokkinos, A.; Ikonomidis, I. Pre-Existing Cytokine and NLRP3 Inflammasome Activation and Increased Vascular Permeability in Diabetes: A Possible Fatal Link with Worst COVID-19 Infection Outcomes? Front. Immunol. 2020, 11, 557235. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J. Oxidative Stress and Endothelial Dysfunction in Sepsis and Acute Inflammation. Antioxid. Redox Signal. 2021, 35, 1291–1307. [Google Scholar] [CrossRef]
- Hu, Z.; Cano, I.; D’Amore, P.A. Update on the role of the endothelial glycocalyx in angiogenesis and vascular inflammation. Front. Cell. Dev. Biol. 2021, 9, 734276. [Google Scholar] [CrossRef]
- Hoogen, I.J.V.D.; Gianni, U.; Wood, M.J.; Taqueti, V.R.; Lin, F.Y.; Hayes, S.N.; Feuchtner, G.M.; van Rosendael, A.R.; Wenger, N.K.; Wei, J.; et al. The Clinical Spectrum of Myocardial Infarction and Ischemia with Nonobstructive Coronary Arteries in Women. JACC Cardiovasc. Imaging 2020, 14, 1053–1062. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Liu, S.; Sun, G.; Sun, Z.; Liu, H. Association of endothelial glycocalyx shedding and coronary microcirculation assessed by an angiography-derived index of microcirculatory resistance in patients with suspected coronary artery disease. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef]
- Pasupathy, S.; Tavella, R.; Grover, S.; Raman, B.; Procter, N.E.; Du, Y.; Mahadavan, G.; Stafford, I.; Heresztyn, T.; Holmes, A.; et al. Early Use of N-acetylcysteine With Nitrate Therapy in Patients Undergoing Primary Percutaneous Coronary Intervention for ST-Segment–Elevation Myocardial Infarction Reduces Myocardial Infarct Size (the NACIAM Trial [N-acetylcysteine in Acute Myocardial Infarction]). Circulation 2017, 136, 894–903. [Google Scholar] [CrossRef]
- Imam, H.; Nguyen, T.H.; Stafford, I.; Liu, S.; Heresztyn, T.; Chirkov, Y.Y.; Horowitz, J.D. Impairment of platelet NO signalling in coronary artery spasm: Role of hydrogen sulphide. Br. J. Pharmacol. 2021, 178, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Falcinelli, E.; Loffredo, F.; Cimmino, G.; Corazzi, T.; Forte, L.; Guglielmini, G.; Momi, S.; Golino, P. Platelets release matrix metalloproteinase-2 in the coronary circulation of patients with acute coronary syndromes: Possible role in sustained platelet activation. Eur. Heart J. 2010, 32, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, M.; Momi, S.; Falcinelli, E.; Bury, L.; Hoylaerts, M.F.; Gresele, P. A novel mechanism regulating human platelet activation by MMP-2–mediated PAR1 biased signaling. Blood 2017, 129, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Brettner, F.; Doerfler, N.; Jacob, M.; Rehm, M.; Bruegger, D.; Conzen, P.; Jacob, B.; Becker, B.F. Protection of glycocalyx decreases platelet adhesion after ischaemia/reperfusion. Eur. J. Anaesthesiol. 2014, 31, 474–481. [Google Scholar] [CrossRef]
- Grushko, O.G.; Cho, S.; Tate, A.M.; Rosenson, R.S.; Pinsky, D.J.; Haus, J.M.; Hummel, S.L.; Goonewardena, S.N. Glycocalyx Disruption Triggers Human Monocyte Activation in Acute Heart Failure Syndromes. Cardiovasc. Drugs Ther. 2022; in press. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2015, 219, 22–96. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Tawa, M.; Okamura, T. Factors influencing the soluble guanylate cyclase heme redox state in blood vessels. Vasc. Pharmacol. 2022, 145, 107023. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Z.; Leung, S.W.; Vanhoutte, P.M. Hypoxic Vasospasm Mediated by cIMP: When Soluble Guanylyl Cyclase Turns Bad. J. Cardiovasc. Pharmacol. 2015, 65, 545–548. [Google Scholar] [CrossRef]
- Chirkov, Y.Y.; Nguyen, T.H.; Horowitz, J.D. Impairment of Anti-Aggregatory Responses to Nitric Oxide and Prostacyclin: Mechanisms and Clinical Implications in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 1042. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, D.; König, A.; Bassenge, E.; Busse, R. Prostacyclin and Nitric Oxide Contribute to the Vasodilator Action of Acetylcholine and Bradykinin in the Intact Rabbit Coronary Bed. J. Cardiovasc. Pharmacol. 1992, 20, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen Sulfide (H(2)S) and Polysulfide (H(2)S(n)) Signaling: The First 25 Years. Biomolecules 2021, 11, 896. [Google Scholar] [CrossRef]
- Bibli, S.-I.; Yang, G.; Zhou, Z.; Wang, R.; Topouzis, S.; Papapetropoulos, A. Role of cGMP in hydrogen sulfide signaling. Nitric. Oxide 2015, 46, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Jeddi, S.; Kashfi, K.; Ghasemi, A. Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension. Biochem. Pharmacol. 2018, 149, 42–59. [Google Scholar] [CrossRef]
- Chatzianastasiou, A.; Bibli, S.-I.; Andreadou, I.; Efentakis, P.; Kaludercic, N.; Wood, M.E.; Whiteman, M.; Di Lisa, F.; Daiber, A.; Manolopoulos, V.G.; et al. Cardioprotection by H2S Donors: Nitric Oxide-Dependent and -Independent Mechanisms. Experiment 2016, 358, 431–440. [Google Scholar] [CrossRef]
- Cao, X.; Wu, Z.; Xiong, S.; Cao, L.; Sethi, G.; Bian, J.-S. The role of hydrogen sulfide in cyclic nucleotide signaling. Biochem. Pharmacol. 2018, 149, 20–28. [Google Scholar] [CrossRef]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Castelblanco, M.; Lugrin, J.; Ehirchiou, D.; Nasi, S.; Ishii, I.; So, A.; Martinon, F.; Busso, N. Hydrogen sulfide inhibits NLRP3 inflammasome activation and reduces cytokine production both in vitro and in a mouse model of inflammation. J. Biol. Chem. 2018, 293, 2546–2557. [Google Scholar] [CrossRef]
- Rajendran, S.; Chirkov, Y.Y.; Campbell, D.J.; Horowitz, J. Angiotensin-(1-7) Enhances Anti-Aggregatory Effects of the Nitric Oxide Donor Sodium Nitroprusside. J. Cardiovasc. Pharmacol. 2005, 46, 459–463. [Google Scholar] [CrossRef]
- Welsh, D.G. The Secret Life of Telomerase. Circ. Res. 2016, 118, 781–782. [Google Scholar] [CrossRef]
- Gutterman, D.G.; Nasci, V.L.; Salato, V.K.; Hijjawi, J.B.; Reuben, C.F.; North, P.E.; Beyer, A.M. Vascular Actions of Angiotensin 1-7 in the Human Microcirculation: Novel Role for Telomerase. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1254–1262. [Google Scholar]
- Delivanis, D.-A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; Kalogeromitros, D. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Ashina, K.; Tsubosaka, Y.; Nakamura, T.; Omori, K.; Kobayashi, K.; Hori, M.; Ozaki, H.; Murata, T. Histamine Induces Vascular Hyperpermeability by Increasing Blood Flow and Endothelial Barrier Disruption In Vivo. PLoS ONE 2015, 10, e0132367. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, S.R.; Stewart, S.; Holmes, A.S.; Chirkov, Y.Y.; Horowitz, J.D. Platelet nitric oxide responsiveness: A novel prognostic marker in acute coronary syndromes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2661–2666. [Google Scholar] [CrossRef]
- Aldiwani, H.; Mahdai, S.; Alhatemi, G.; Merz, C.N.B. Microvascular Angina: Diagnosis and Management. Eur. Cardiol. Rev. 2021, 16, e46. [Google Scholar] [CrossRef]
- Ford, T.; Ong, P.; Sechtem, U.; Beltrame, J.; Camici, P.G.; Crea, F.; Kaski, J.-C.; Merz, C.N.B.; Pepine, C.J.; Shimokawa, H.; et al. Assessment of Vascular Dysfunction in Patients Without Obstructive Coronary Artery Disease. JACC Cardiovasc. Interv. 2020, 13, 1847–1864. [Google Scholar] [CrossRef]
- Aziz, A.; Hansen, H.S.; Sechtem, U.; Prescott, E.; Ong, P. Sex-Related Differences in Vasomotor Function in Patients With Angina and Unobstructed Coronary Arteries. J. Am. Coll. Cardiol. 2017, 70, 2349–2358. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Diaz, A.; Cyr, D.D.; Shaw, L.J.; Mancini, G.B.J.; Leipsic, J.; Budoff, M.J.; Min, J.K.; Hague, C.J.; Berman, D.S.; et al. Ischemia with Nonobstructive Coronary Arteries: Insights from the ISCHEMIA Trial. JACC Cardiovasc. Imaging 2023, 16, 63–74. [Google Scholar]
- Chirkov, Y.Y.; Chirkova, L.P.; Sage, R.E.; Horowitz, J.D. Impaired responsiveness of platelets from patients with stable angina pectoris to antiaggregating and cyclicAMP-elevating effects of prostaglandin E1. J. Cardiovasc. Pharmacol. 1995, 25, 961–966. [Google Scholar] [CrossRef]
- Chirkov, Y.Y.; Holmes, A.S.; Chirkova, L.P.; Horowitz, J.D. Nitrate Resistance in Platelets From Patients With Stable Angina Pectoris. Circulation 1999, 100, 129–134. [Google Scholar] [CrossRef]
- Prinzmetal, M.; Kennamer, R.; Merliss, R.; Wada, T.; Bor, N. Angina pectoris. I. A variant form of angina pectoris; preliminary report. Am. J. Med. 1959, 27, 375–388. [Google Scholar] [CrossRef]
- Beltrame, J.F.; Limaye, S.B.; Horowitz, J.D. The Coronary Slow Flow Phenomenon—A New Coronary Microvascular Disorder. Cardiology 2002, 97, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, J.F.; Turner, S.P.; Leslie, S.L.; Solomon, P.; Freedman, S.B.; Horowitz, J.D. The angiographic and clinical benefits of mibefradil in the coronary slow flow phenomenon. J. Am. Coll. Cardiol. 2004, 44, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Razgonova, M.P.; Zakharenko, A.M.; Golokhvast, K.S.; Thanasoula, M.; Sarandi, E.; Nikolouzakis, K.; Fragkiadaki, P.; Tsoukalas, D.; Spandidos, D.A.; Tsatsakis, A. Telomerase and telomeres in aging theory and chronographic aging theory (Review). Mol. Med. Rep. 2020, 22, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.M.; Freed, J.K.; Durand, M.J.; Riedel, M.; Ait-Aissa, K.; Green, P.; Hockenberry, J.C.; Morgan, R.G.; Donato, A.J.; Peleg, R.; et al. Critical Role for Telomerase in the Mechanism of Flow-Mediated Dilation in the Human Microcirculation. Circ. Res. 2016, 118, 856–866. [Google Scholar] [CrossRef]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef]
- Vervloet, M.G.; Adema, A.Y.; Larsson, T.E.; Massy, Z.A. The Role of Klotho on Vascular Calcification and Endothelial Function in Chronic Kidney Disease. Semin. Nephrol. 2014, 34, 578–585. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Z.-H.; Ren, Z.; Qu, S.-L.; Liu, M.-H.; Liu, L.-S.; Jiang, Z.-S. Hydrogen Sulfide, the Next Potent Preventive and Therapeutic Agent in Aging and Age-Associated Diseases. Mol. Cell. Biol. 2013, 33, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Chirkov, Y.Y.; Horowitz, J.D. Aging of the nitric oxide system: Are we as old as our NO? J. Am. Heart Assoc. 2014, 3, e000973. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.; Chan, W.P.; Procter, N.E.; Chirkov, Y.Y.; Ngo, D.T.; Horowitz, J. Reciprocal regulation of NO signaling and TXNIP expression in humans: Impact of aging and ramipril therapy. Int. J. Cardiol. 2013, 168, 4624–4630. [Google Scholar] [CrossRef]
- Shi, Y.; Feletou, M.; Ku, D.D.; Man, R.Y.K.; Vanhoutte, P.M. The calcium ionophore A23187 induces endothelium-dependent contractions in femoral arteries from rats with streptozotocin-induced diabetes. Br. J. Pharmacol. 2007, 150, 624–632. [Google Scholar] [CrossRef]
- Williams, S.B.; Cusco, J.A.; Roddy, M.-A.; Johnstone, M.T.; Creager, M.A. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1996, 27, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Kucia, A.M.; Heresztyn, T.; Stewart, S.; Chirkov, Y.Y.; Zeitz, C.J.; Horowitz, J.D. The deleterious effects of hyperglycemia on platelet function in diabetic patients with acute coronary syndromes mediation by superoxide production, resolution with intensive insulin administration. J. Am. Coll. Cardiol. 2007, 49, 304–310. [Google Scholar]
- Dogne, S.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar] [CrossRef]
- Chiribiri, A.; Masci, P.G. From the Epicardial Vessels to the Microcirculation: The Coronary Vasculature at the Crossroad of HFpEF. JACC Cardiovasc. Imaging 2021, 14, 2334–2336. [Google Scholar]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A vascular disease. Trends Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e8. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-Dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Mongelli, A.; Barbi, V.; Zamperla, M.G.; Atlante, S.; Forleo, L.; Nesta, M.; Massetti, M.; Pontecorvi, A.; Nanni, S.; Farsetti, A.; et al. Evidence for Biological Age Acceleration and Telomere Shortening in COVID-19 Survivors. Int. J. Mol. Sci. 2021, 22, 6151. [Google Scholar] [CrossRef]
- Ramchand, J.; Burrell, L.M. Circulating ACE2: A novel biomarker of cardiovascular risk. Lancet 2020, 396, 937–939. [Google Scholar] [CrossRef]
- Narula, S.; Yusuf, S.; Chong, M.; Ramasundarahettige, C.; Rangarajan, S.; I Bangdiwala, S.; van Eikels, M.; Leineweber, K.; Wu, A.; Pigeyre, M.; et al. Plasma ACE2 and risk of death or cardiometabolic diseases: A case-cohort analysis. Lancet 2020, 396, 968–976. [Google Scholar] [CrossRef]
- Giustarini, D.; Rossi, R.; Kukoc-Modun, L.; Kedia, G.; Uckert, S. S-Nitroso-N-acetyl-L-cysteine ethyl ester (SNACET) and N-acetyl-L-cysteine ethyl ester (NACET)-Cysteine-based drug candidates with unique pharmacological profiles for oral use as, NO.; H(2)S and GSH suppliers and as antioxidants: Results and overview. J. Pharm. Anal. 2018, 8, 1–9. [Google Scholar]
- Elies, J.; Scragg, J.L.; Huang, S.; Dallas, M.L.; Huang, D.; MacDougall, D.; Boyle, J.P.; Gamper, N.; Peers, C. Hydrogen sulfide inhibits Cav3.2 T-type Ca 2+ channels. FASEB J. 2014, 28, 5376–5387. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Juni, R.P.; Kuster, D.W.; Goebel, M.; Helmes, M.; Musters, R.J.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, C. The cystathionine-γ-lyase/hydrogen sulfide pathway mediates the trimetazidine-iinduced protection of H9c2 cells against hypoxia/reoxygenation-induced apoptosis and oxidative stress. Anatol. J. Cardiol. 2019, 22, 102–111. [Google Scholar] [PubMed]
- Zhang, T.; Finn, D.F.; Barlow, J.W.; Walsh, J.J. Mast cell stabilizers. Eur. J. Pharmacol. 2016, 778, 158–168. [Google Scholar] [PubMed]
- Campeau, M.-A.; Leask, R.L. Empagliflozin mitigates endothelial inflammation and attenuates endoplasmic reticulum stress signaling caused by sustained glycocalyx disruption. Sci. Rep. 2022, 12, 12681. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Event (n = 2591) | Conservative Group (n = 2591) | Invasive Group (n = 2588) |
|---|---|---|
| Primary endpoint | 451 | 420 |
| Death | 144 | 145 |
| Myocardial infarction | 268 | 244 |
| Heart failure | 33 | 62 |
| Unstable angina | 35 | 17 |
| Clinical Parameter | p-Value | Odd Ratio |
|---|---|---|
| Increasing age | <0.001 | 0.77 |
| Female | <0.01 | 4.19 |
| Diabetes | <0.01 | 0.56 |
| Cigarette smoking | NS | 0.68 |
| Category | Examples | Potential Clinical Utility |
|---|---|---|
Mast cell stabilisation:
| Nitric oxide (NO), prostacyclin (PGI2) Hydrogen sulphide(H2S) Corticosteroids Trimetazidine | Both NO and PGI2 may inhibit mast cell activation and degranulation. However, no clinical studies to date have conclusively shown the benefit of exogenous administration of such agents. The H2S donor N-acetylcysteine, in association with infused nitroglycerine, has been shown to reduce myocardial infarct size [50] and to stabilise crises of coronary artery spasm [51], with associated reduction in plasma tryptase concentrations. There are isolated case reports documenting reversal of coronary spasm crises with high-dose corticosteroid “pulse” therapy. Trimetazidine, which is known to act as a partial fatty acid oxidation inhibitor, has an established antianginal effect. This may be mediated, in part, by increased formation of H2S [114]. |
| Inhibition of “sheddase” (largely matrix metalloproteinase) effect in degrading glycocalyx | (Low-dose) Doxycycline | Low-dose doxycycline, while exerting no antibiotic effect, acts as a nonspecific inhibitor of matrix metalloproteinases. However, no trials to date have convincingly established its utility to prevent coronary events. A number of other candidate MMP inhibitors are under development [115]. |
| Increased glycocalyx regeneration | Albumin | Albumin is the major plasma protein and is responsible for the majority of plasma oncotic pressure. In vitro studies have shown that glycocalyx regeneration, especially in diabetic models, is accelerated by albumin. However, plasma albumin repletion has not been utilised therapeutically to date. |
| Reversal of endothelial inflammation | SGLT2 inhibitors | Two studies [112,116] have shown inhibition of microvascular inflammation by SGLT2 inhibitors. A similar effect was seen with N-acetylcysteine [116], but full mechanism(s) is not yet delineated. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kei, C.Y.; Singh, K.; Dautov, R.F.; Nguyen, T.H.; Chirkov, Y.Y.; Horowitz, J.D. Coronary “Microvascular Dysfunction”: Evolving Understanding of Pathophysiology, Clinical Implications, and Potential Therapeutics. Int. J. Mol. Sci. 2023, 24, 11287. https://doi.org/10.3390/ijms241411287
Kei CY, Singh K, Dautov RF, Nguyen TH, Chirkov YY, Horowitz JD. Coronary “Microvascular Dysfunction”: Evolving Understanding of Pathophysiology, Clinical Implications, and Potential Therapeutics. International Journal of Molecular Sciences. 2023; 24(14):11287. https://doi.org/10.3390/ijms241411287
Chicago/Turabian StyleKei, Chun Yeung, Kuljit Singh, Rustem F. Dautov, Thanh H. Nguyen, Yuliy Y. Chirkov, and John D. Horowitz. 2023. "Coronary “Microvascular Dysfunction”: Evolving Understanding of Pathophysiology, Clinical Implications, and Potential Therapeutics" International Journal of Molecular Sciences 24, no. 14: 11287. https://doi.org/10.3390/ijms241411287
APA StyleKei, C. Y., Singh, K., Dautov, R. F., Nguyen, T. H., Chirkov, Y. Y., & Horowitz, J. D. (2023). Coronary “Microvascular Dysfunction”: Evolving Understanding of Pathophysiology, Clinical Implications, and Potential Therapeutics. International Journal of Molecular Sciences, 24(14), 11287. https://doi.org/10.3390/ijms241411287