

Effects of a True Prophylactic Treatment on Hippocampal and Amygdala Synaptic Plasticity and Gene Expression in a Rodent Chronic Stress Model of Social Defeat

 , , ,
, , ,  , , , ,
, , , , 
Abstract
1. Introduction
2. Results
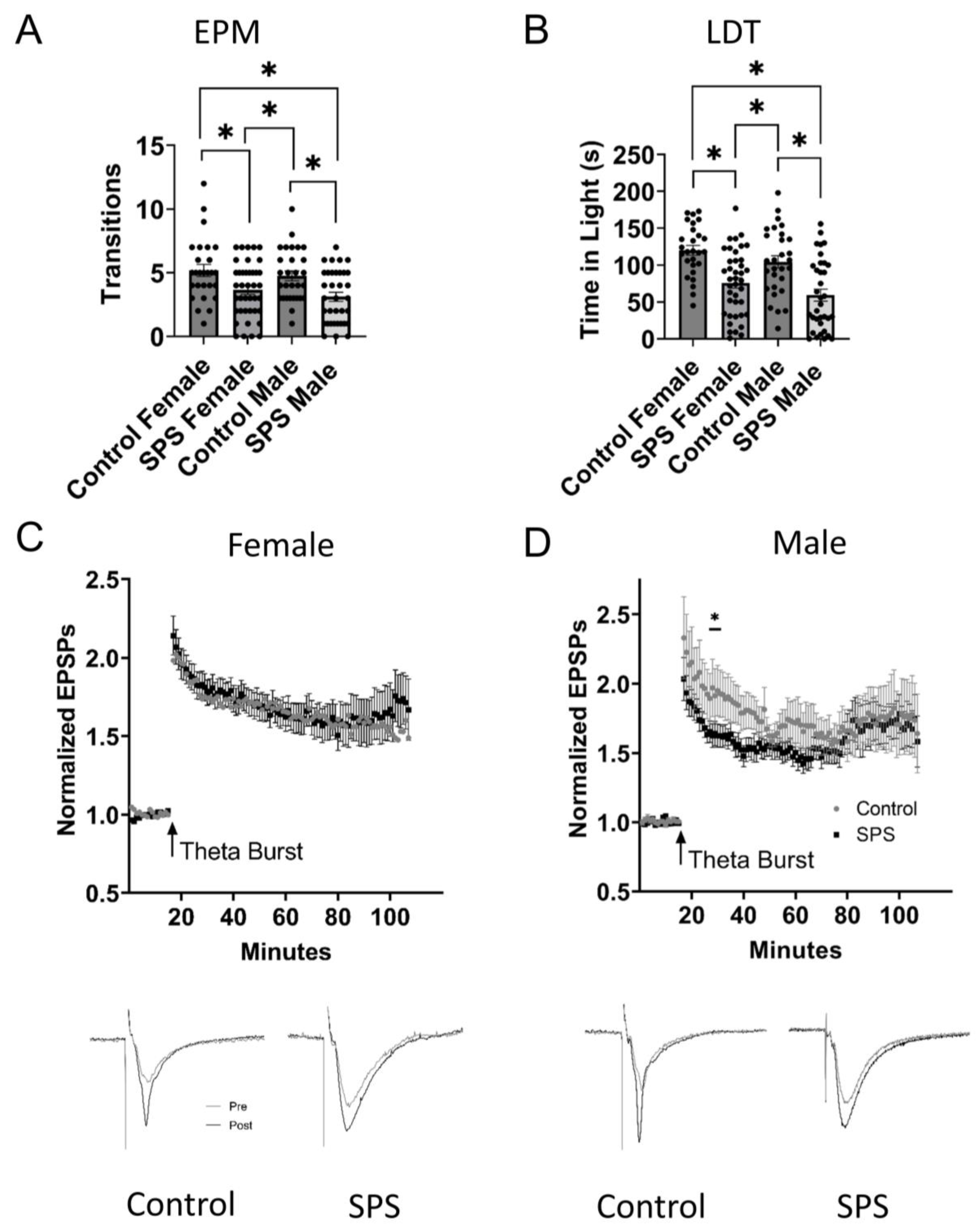
2.1. Male and Female Singled Prolonged Stress (SPS)
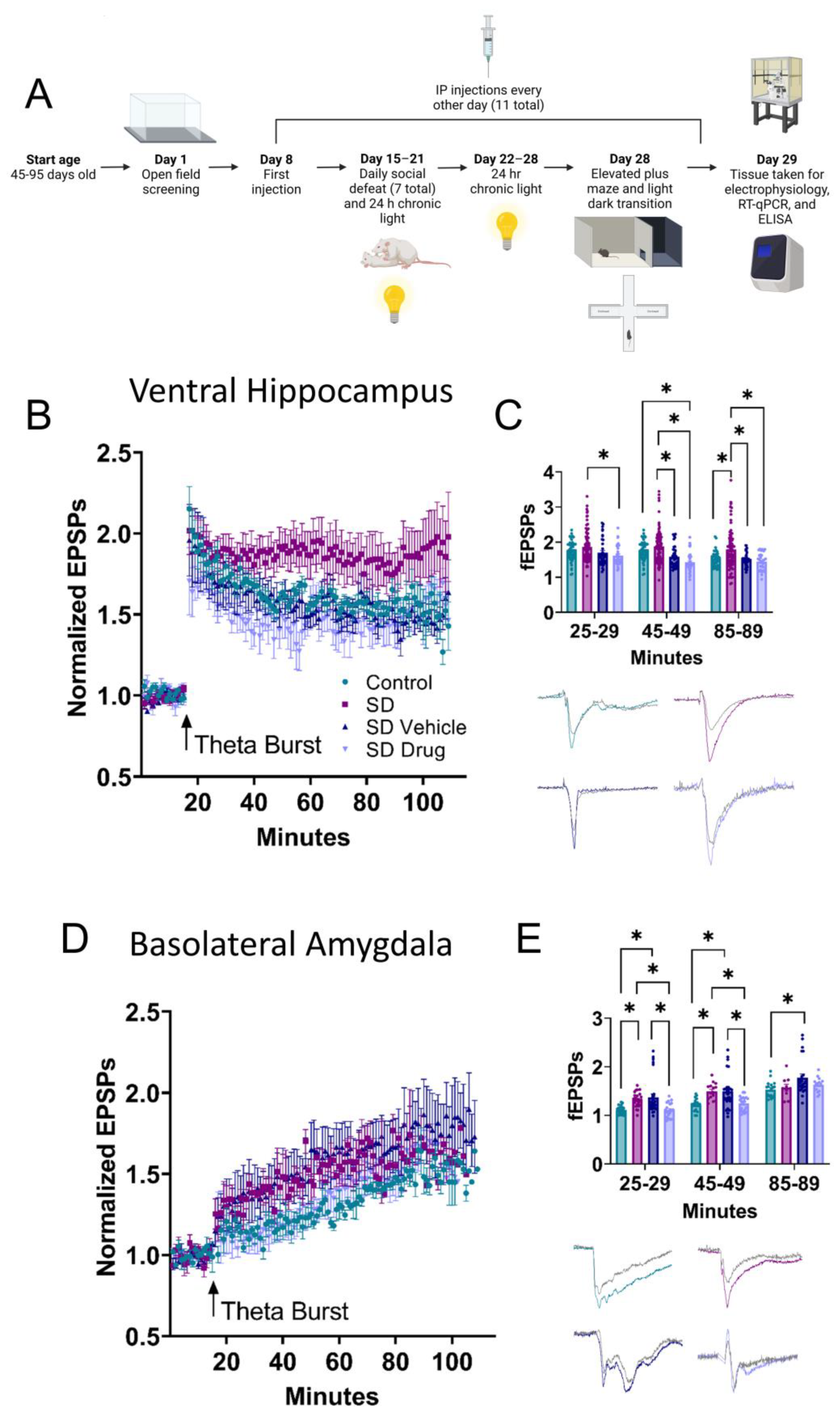
2.2. Ventral Hippocampal and Basolateral Amygdala Field Electrophysiology
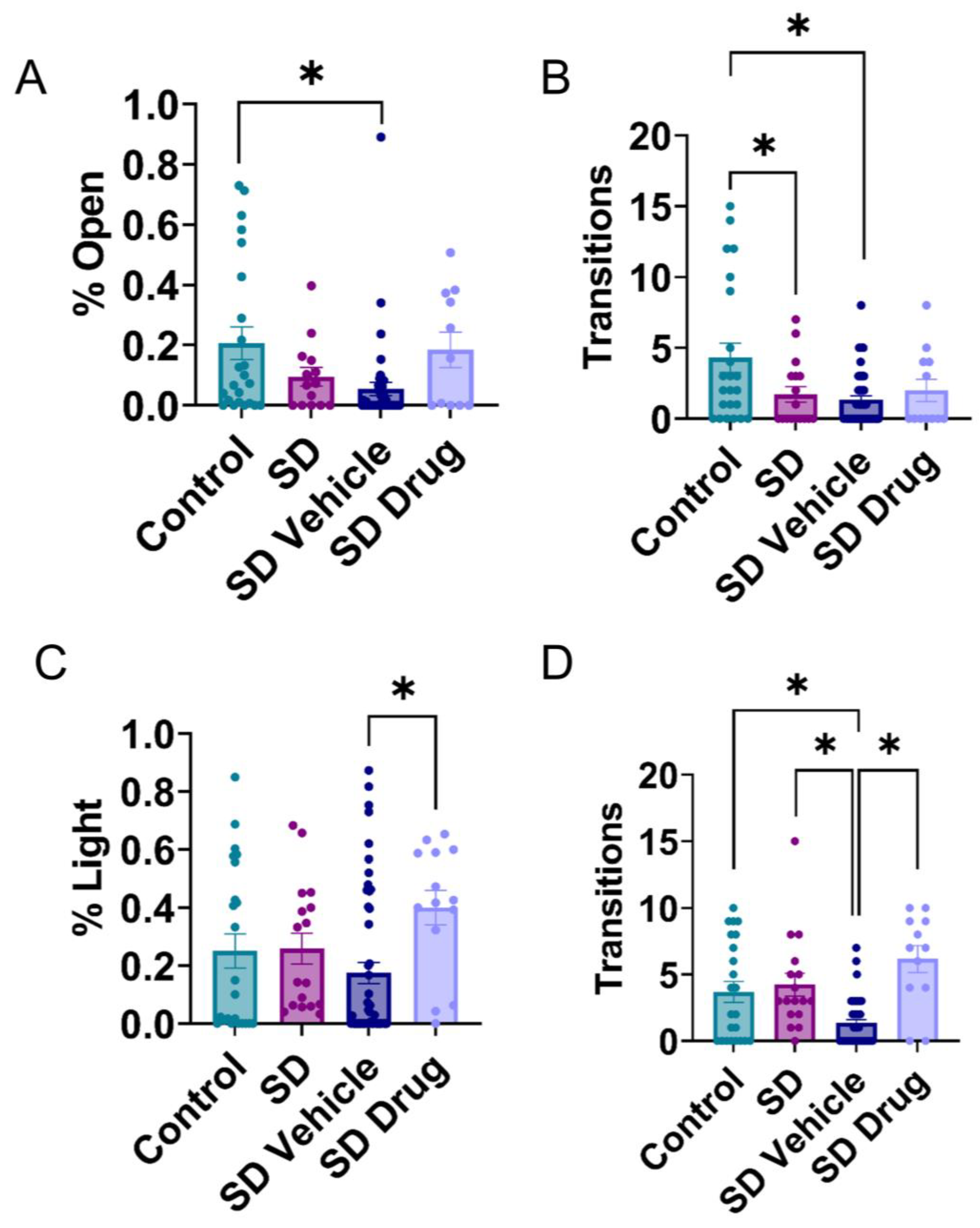
2.3. Behavioral Testing in the Elevated plus Maze and Light-Dark Transition Test
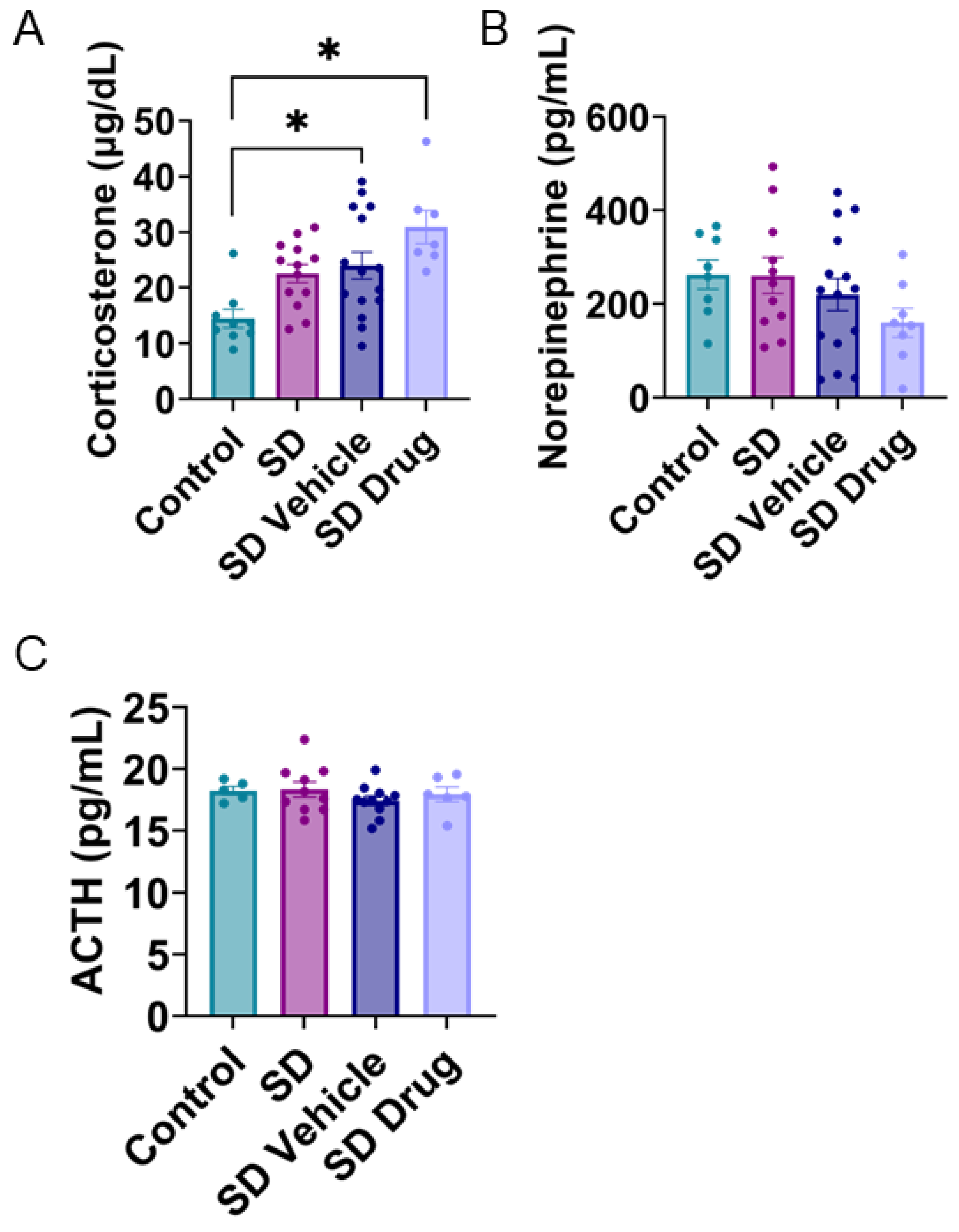
2.4. Plasma Corticosterone, Norepinephrine, and ACTH Concentrations
2.5. Relative mRNA Expression Levels of Post-Chronic Stress-Related Target Genes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Screening Protocol
4.3. Single Prolonged Stress
4.4. Social Defeat
4.5. Drug Preparation and Administration
4.6. Elevated plus Maze (EPM)
4.7. Light–Dark Transition (LDT)
4.8. Brain Slice Preparation
4.9. Field Electrophysiology
4.10. Tissue Extraction and Reverse Transcription Quantitative PCR (RT-qPCR)
4.11. Competitive Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, M.J. Posttraumatic stress disorder. J. Clin. Psychiatry 1997, 58 (Suppl. S9), 33–36. [Google Scholar]
- Vieweg, W.V.; Julius, D.A.; Fernandez, A.; Beatty-Brooks, M.; Hettema, J.M.; Pandurangi, A.K. Posttraumatic stress disorder: Clinical features, pathophysiology, and treatment. Am. J. Med. 2006, 119, 383–390. [Google Scholar] [CrossRef]
- Williamson, J.B.; Jaffee, M.S.; Jorge, R.E. Posttraumatic Stress Disorder and Anxiety-Related Conditions. Continuum 2021, 27, 1738–1763. [Google Scholar] [CrossRef]
- Shin, L.M.; Rauch, S.L.; Pitman, R.K. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. In Psychobiology of Posttraumatic Stress Disorder: A Decade of Progress; Wiley-Blackwell: Hoboken, NJ, USA, 2006; Volume 1071, pp. 67–79. [Google Scholar]
- Mahan, A.L.; Ressler, K.J. Fear conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef]
- Moustafa, A.A.; Gilbertson, M.W.; Orr, S.P.; Herzallah, M.M.; Servatius, R.J.; Myers, C.E. A model of amygdala-hippocampal-prefrontal interaction in fear conditioning and extinction in animals. Brain Cogn. 2013, 81, 29–43. [Google Scholar] [CrossRef]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef]
- Goto, A.; Bota, A.; Miya, K.; Wang, J.; Tsukamoto, S.; Jiang, X.; Hirai, D.; Murayama, M.; Matsuda, T.; McHugh, T.J.; et al. Stepwise synaptic plasticity events drive the early phase of memory consolidation. Science 2021, 374, 857–863. [Google Scholar] [CrossRef]
- Howland, J.G.; Wang, Y.T. Synaptic plasticity in learning and memory: Stress effects in the hippocampus. Prog. Brain Res. 2008, 169, 145–158. [Google Scholar] [CrossRef]
- Kim, J.J.; Song, E.Y.; Kosten, T.A. Stress effects in the hippocampus: Synaptic plasticity and memory. Stress 2006, 9, 1–11. [Google Scholar] [CrossRef]
- Keralapurath, M.M.; Clark, J.K.; Hammond, S.; Wagner, J.J. Cocaine- or stress-induced metaplasticity of LTP in the dorsal and ventral hippocampus. Hippocampus 2014, 24, 577–590. [Google Scholar] [CrossRef]
- McKernan, M.G.; Shinnick-Gallagher, P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 1997, 390, 607–611. [Google Scholar] [CrossRef]
- Pape, H.C.; Pare, D. Plastic Synaptic Networks of the Amygdala for the Acquisition, Expression, and Extinction of Conditioned Fear. Physiol. Rev. 2010, 90, 419–463. [Google Scholar] [CrossRef]
- Rogan, M.T.; Stäubli, U.V.; LeDoux, J.E. Fear conditioning induces associative long-term potentiation in the amygdala. Nature 1997, 390, 604–607. [Google Scholar] [CrossRef]
- Sah, P.; Westbrook, R.F.; Lüthi, A. Fear conditioning and long-term potentiation in the amygdala: What really is the connection? Ann. N. Y. Acad. Sci. 2008, 1129, 88–95. [Google Scholar] [CrossRef]
- Song, C.; Detert, J.A.; Sehgal, M.; Moyer, J.R., Jr. Trace fear conditioning enhances synaptic and intrinsic plasticity in rat hippocampus. J. Neurophysiol. 2012, 107, 3397–3408. [Google Scholar] [CrossRef]
- Francati, V.; Vermetten, E.; Bremner, J.D. Functional neuroimaging studies in posttraumatic stress disorder: Review of current methods and findings. Depress. Anxiety 2007, 24, 202–218. [Google Scholar] [CrossRef]
- Pu, Z.; Krugers, H.J.; Joels, M. Corticosterone time-dependently modulates beta-adrenergic effects on long-term potentiation in the hippocampal dentate gyrus. Learn. Mem. 2007, 14, 359–367. [Google Scholar] [CrossRef]
- Sarabdjitsingh, R.A.; Kofink, D.; Karst, H.; de Kloet, E.R.; Joëls, M. Stress-induced enhancement of mouse amygdalar synaptic plasticity depends on glucocorticoid and ß-adrenergic activity. PLoS ONE 2012, 7, e42143. [Google Scholar] [CrossRef]
- Grigoryan, G.; Ardi, Z.; Albrecht, A.; Richter-Levin, G.; Segal, M. Juvenile stress alters LTP in ventral hippocampal slices: Involvement of noradrenergic mechanisms. Behav. Brain Res. 2015, 278, 559–562. [Google Scholar] [CrossRef]
- Debiec, J.; Bush, D.E.A.; LeDoux, J.E. Noradrenergic Enhancement of Reconsolidation in the Amygdala Impairs Extinction of Conditioned Fear in Rats—A Possible Mechanism for the Persistence of Traumatic Memories in PTSD. Depress. Anxiety 2011, 28, 186–193. [Google Scholar] [CrossRef]
- Pitman, R.K.; Milad, M.R.; Igoe, S.A.; Vangel, M.G.; Orr, S.P.; Tsareva, A.; Gamache, K.; Nader, K. Systemic mifepristone blocks reconsolidation of cue-conditioned fear; propranolol prevents this effect. Behav. Neurosci. 2011, 125, 632–638. [Google Scholar] [CrossRef]
- Lin, C.C.; Cheng, P.Y.; Hsiao, M.; Liu, Y.P. Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. Int. J. Mol. Sci. 2022, 23, 5494. [Google Scholar] [CrossRef]
- Calfa, G.; Bussolino, D.; Molina, V.A. Involvement of the lateral septum and the ventral Hippocampus in the emotional sequelae induced by social defeat: Role of glucocorticoid receptors. Behav. Brain Res. 2007, 181, 23–34. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef]
- Wang, W.; Liu, L.; Yang, X.; Gao, H.; Tang, Q.K.; Yin, L.Y.; Yin, X.Y.; Hao, J.R.; Geng, D.Q.; Gao, C. Ketamine improved depressive-like behaviors via hippocampal glucocorticoid receptor in chronic stress induced-susceptible mice. Behav. Brain Res. 2019, 364, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Mouri, A.; Ukai, M.; Uchida, M.; Hasegawa, S.; Taniguchi, M.; Ito, T.; Hida, H.; Yoshimi, A.; Yamada, K.; Kunimoto, S.; et al. Juvenile social defeat stress exposure persistently impairs social behaviors and neurogenesis. Neuropharmacology 2018, 133, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.; Huang, C.C.; Hsu, K.S. Brain-derived neurotrophic factor in the amygdala mediates susceptibility to fear conditioning. Exp. Neurol. 2014, 255, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Burke, A.R.; Renner, K.J.; Forster, G.L. Adolescent male rats exposed to social defeat exhibit altered anxiety behavior and limbic monoamines as adults. Behav. Neurosci. 2009, 123, 564–576. [Google Scholar] [CrossRef]
- Parise, L.F.; Parise, E.M.; Sial, O.K.; Bolaños-Guzmán, C.A. Social Buffering is Dependent on Mutual Experience in Adolescent Male Mice Exposed to Social Defeat Stress. Chronic Stress 2022, 6, 24705470221111094. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, D.; Brennan, K.C. The Effects of Chronic Stress on Migraine Relevant Phenotypes in Male Mice. Front. Cell Neurosci. 2018, 12, 294. [Google Scholar] [CrossRef]
- Lu, Q.; Xiang, H.; Zhu, H.; Chen, Y.; Lu, X.; Huang, C. Intranasal lipopolysaccharide administration prevents chronic stress-induced depression- and anxiety-like behaviors in mice. Neuropharmacology 2021, 200, 108816. [Google Scholar] [CrossRef]
- Deslauriers, J.; Toth, M.; Der-Avakian, A.; Risbrough, V.B. Current Status of Animal Models of Posttraumatic Stress Disorder: Behavioral and Biological Phenotypes, and Future Challenges in Improving Translation. Biol. Psychiatry 2018, 83, 895–907. [Google Scholar] [CrossRef]
- Flandreau, E.I.; Toth, M. Animal Models of PTSD: A Critical Review. In Behavioral Neurobiology of PTSD; Vermetten, E., Baker, D.G., Risbrough, V.B., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 47–68. [Google Scholar] [CrossRef]
- Schöner, J.; Heinz, A.; Endres, M.; Gertz, K.; Kronenberg, G. Post-traumatic stress disorder and beyond: An overview of rodent stress models. J. Cell Mol. Med. 2017, 21, 2248–2256. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, X.-Z.; Li, H.; Li, X.; Yu, T.; Dohl, J.; Ursano, R.J. Updates in PTSD Animal Models Characterization. In Psychiatric Disorders: Methods and Protocols; Kobeissy, F.H., Ed.; Springer: New York, NY, USA, 2019; pp. 331–344. [Google Scholar] [CrossRef]
- Bedrosian, T.A.; Fonken, L.K.; Walton, J.C.; Haim, A.; Nelson, R.J. Dim light at night provokes depression-like behaviors and reduces CA1 dendritic spine density in female hamsters. Psychoneuroendocrinology 2011, 36, 1062–1069. [Google Scholar] [CrossRef]
- Richter-Levin, G.; Sandi, C. Title: "Labels Matter: Is it stress or is it Trauma?". Transl. Psychiatry 2021, 11, 385. [Google Scholar] [CrossRef]
- Golden, S.A.; Covington, H.E., 3rd; Berton, O.; Russo, S.J. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011, 6, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Koolhaas, J.M.; de Boer, S.F.; Buwalda, B.; Meerlo, P. Social stress models in rodents: Towards enhanced validity. Neurobiol. Stress 2017, 6, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Koolhaas, J.M.; Korte, S.M.; De Boer, S.F.; Van Der Vegt, B.J.; Van Reenen, C.G.; Hopster, H.; De Jong, I.C.; Ruis, M.A.; Blokhuis, H.J. Coping styles in animals: Current status in behavior and stress-physiology. Neurosci. Biobehav. Rev. 1999, 23, 925–935. [Google Scholar] [CrossRef]
- de Boer, S.F.; Buwalda, B.; Koolhaas, J.M. Untangling the neurobiology of coping styles in rodents: Towards neural mechanisms underlying individual differences in disease susceptibility. Neurosci. Biobehav. Rev. 2017, 74, 401–422. [Google Scholar] [CrossRef]
- Koolhaas, J.M.; de Boer, S.F.; Buwalda, B.; van Reenen, K. Individual variation in coping with stress: A multidimensional approach of ultimate and proximate mechanisms. Brain Behav. Evol. 2007, 70, 218–226. [Google Scholar] [CrossRef] [PubMed]
- LeDoux, J.E.; Gorman, J.M. A call to action: Overcoming anxiety through active coping. Am. J. Psychiatry 2001, 158, 1953–1955. [Google Scholar] [CrossRef] [PubMed]
- Chiavarino, C.; Rabellino, D.; Ardito, R.B.; Cavallero, E.; Palumbo, L.; Bergerone, S.; Gaita, F.; Bara, B.G. Emotional coping is a better predictor of cardiac prognosis than depression and anxiety. J. Psychosom. Res. 2012, 73, 473–475. [Google Scholar] [CrossRef]
- Russo, S.J.; Murrough, J.W.; Han, M.H.; Charney, D.S.; Nestler, E.J. Neurobiology of resilience. Nat. Neurosci. 2012, 15, 1475–1484. [Google Scholar] [CrossRef]
- Veenema, A.H.; Neumann, I.D. Neurobiological mechanisms of aggression and stress coping: A comparative study in mouse and rat selection lines. Brain Behav. Evol. 2007, 70, 274–285. [Google Scholar] [CrossRef]
- Kubo, K.Y.; Iinuma, M.; Chen, H. Mastication as a Stress-Coping Behavior. BioMed Res. Int. 2015, 2015, 876409. [Google Scholar] [CrossRef] [PubMed]
- Steinman, M.Q.; Trainor, B.C. Sex differences in the effects of social defeat on brain and behavior in the California mouse: Insights from a monogamous rodent. Semin. Cell Dev. Biol. 2017, 61, 92–98. [Google Scholar] [CrossRef]
- Maren, S. Long-term potentiation in the amygdala: A mechanism for emotional learning and memory. Trends Neurosci. 1999, 22, 561–567. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, H.J.; Han, J.S.; Packard, M.G. Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J. Neurosci. 2001, 21, 5222–5228. [Google Scholar] [CrossRef]
- Elzinga, B.M.; Bremner, J.D. Are the neural substrates of memory the final common pathway in posttraumatic stress disorder (PTSD)? J. Affect. Disord. 2002, 70, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.M.; Campbell, A.M.; Park, C.R.; Halonen, J.; Zoladz, P.R. The Temporal Dynamics Model of Emotional Memory Processing: A Synthesis on the Neurobiological Basis of Stress-Induced Amnesia, Flashbulb and Traumatic Memories, and the Yerkes-Dodson Law. Neural Plast. 2007, 2007, 060803. [Google Scholar] [CrossRef]
- Jarrard, L.E. On the role of the hippocampus in learning and memory in the rat. Behav. Neural. Biol. 1993, 60, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Unique regulation of long term potentiation in the rat ventral hippocampus. Hippocampus 2007, 17, 10–25. [Google Scholar] [CrossRef]
- Suvrathan, A.; Bennur, S.; Ghosh, S.; Tomar, A.; Anilkumar, S.; Chattarji, S. Stress enhances fear by forming new synapses with greater capacity for long-term potentiation in the amygdala. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 369, 20130151. [Google Scholar] [CrossRef]
- McDonald, A.J.; Mott, D.D. Functional neuroanatomy of amygdalohippocampal interconnections and their role in learning and memory. J. Neurosci. Res. 2017, 95, 797–820. [Google Scholar] [CrossRef]
- Tank, A.W.; Lee Wong, D. Peripheral and central effects of circulating catecholamines. Compr. Physiol. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joëls, M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 2011, 209, 153–167. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Oitzl, M.S.; Joëls, M. Functional implications of brain corticosteroid receptor diversity. Cell. Mol. Neurobiol. 1993, 13, 433–455. [Google Scholar] [CrossRef]
- Zhe, D.; Fang, H.; Yuxiu, S. Expressions of hippocampal mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) in the single-prolonged stress-rats. Acta Histochem. Cytochem. 2008, 41, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Ding, J.; Shi, Y. Expression of amygdala mineralocorticoid receptor and glucocorticoid receptor in the single-prolonged stress rats. BMC Neurosci. 2014, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Covington, H.E., 3rd; Miczek, K.A. Intense cocaine self-administration after episodic social defeat stress, but not after aggressive behavior: Dissociation from corticosterone activation. Psychopharmacology 2005, 183, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Buwalda, B.; Scholte, J.; de Boer, S.F.; Coppens, C.M.; Koolhaas, J.M. The acute glucocorticoid stress response does not differentiate between rewarding and aversive social stimuli in rats. Horm. Behav. 2012, 61, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.M.; Bennett, M.C.; Fleshner, M.; Rose, G.M. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 1992, 2, 421–430. [Google Scholar] [CrossRef]
- Salehi, B.; Cordero, M.I.; Sandi, C. Learning under stress: The inverted-U-shape function revisited. Learn. Mem. 2010, 17, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.M.; Kölsch, M.; Larra, M.F.; Zech, C.M.; Blumenthal, T.D.; Frings, C.; Schächinger, H. For whom the bell (curve) tolls: Cortisol rapidly affects memory retrieval by an inverted U-shaped dose-response relationship. Psychoneuroendocrinology 2013, 38, 1565–1572. [Google Scholar] [CrossRef]
- Fleseriu, M. Medical treatment of Cushing disease: New targets, new hope. Endocrinol. Metab. Clin. N. Am. 2015, 44, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.C.; Davies, D.R.; Scholl, J.L.; Watt, M.J.; Forster, G.L. Differential effects of glucocorticoid and mineralocorticoid antagonism on anxiety behavior in mild traumatic brain injury. Behav. Brain Res. 2016, 312, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Kohda, K.; Harada, K.; Kato, K.; Hoshino, A.; Motohashi, J.; Yamaji, T.; Morinobu, S.; Matsuoka, N.; Kato, N. Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: A putative post-traumatic stress disorder model. Neuroscience 2007, 148, 22–33. [Google Scholar] [CrossRef]
- Miller, R.M.; Marriott, D.; Trotter, J.; Hammond, T.; Lyman, D.; Call, T.; Walker, B.; Christensen, N.; Haynie, D.; Badura, Z.; et al. Running Exercise Mitigates the Negative Consequences of Chronic Stress on Dorsal Hippocampal Long-Term Potentiation in Male Mice. Neurobiol. Learn. Mem. 2018, 149, 28–38. [Google Scholar] [CrossRef]
- Yang, H.; de Jong, J.W.; Tak, Y.; Peck, J.; Bateup, H.S.; Lammel, S. Nucleus Accumbens Subnuclei Regulate Motivated Behavior via Direct Inhibition and Disinhibition of VTA Dopamine Subpopulations. Neuron 2018, 97, 434–449.e434. [Google Scholar] [CrossRef]
- Sanz-García, A.; Knafo, S.; Pereda-Pérez, I.; Esteban, J.A.; Venero, C.; Armario, A. Administration of the TrkB receptor agonist 7,8-dihydroxyflavone prevents traumatic stress-induced spatial memory deficits and changes in synaptic plasticity. Hippocampus 2016, 26, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Henley, J.M.; Holman, D.; Zhou, M.; Wiegert, O.; van Spronsen, M.; Joels, M.; Hoogenraad, C.C.; Krugers, H.J. Corticosterone Alters AMPAR Mobility and Facilitates Bidirectional Synaptic Plasticity. PLoS ONE 2009, 4, e4714. [Google Scholar] [CrossRef]
- Groc, L.; Choquet, D.; Chaouloff, F. The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat. Neurosci. 2008, 11, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Real, E.; Takamiya, K.; Kang, M.G.; Ledoux, J.; Huganir, R.L.; Malinow, R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 2007, 131, 160–173. [Google Scholar] [CrossRef]
- Krishnan, V.; Han, M.H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; Laplant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef]
- Nalloor, R.; Bunting, K.; Vazdarjanova, A. Predicting impaired extinction of traumatic memory and elevated startle. PLoS ONE 2011, 6, e19760. [Google Scholar] [CrossRef]
- Wernecke, K.E.; Vincenz, D.; Storsberg, S.; D’Hanis, W.; Goldschmidt, J.; Fendt, M. Fox urine exposure induces avoidance behavior in rats and activates the amygdalar olfactory cortex. Behav. Brain Res. 2015, 279, 76–81. [Google Scholar] [CrossRef]
- Atsak, P.; Orre, M.; Bakker, P.; Cerliani, L.; Roozendaal, B.; Gazzola, V.; Moita, M.; Keysers, C. Experience modulates vicarious freezing in rats: A model for empathy. PLoS ONE 2011, 6, e21855. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, K. Freeze for action: Neurobiological mechanisms in animal and human freezing. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372, 20160206. [Google Scholar] [CrossRef]
- Biedermann, S.V.; Biedermann, D.G.; Wenzlaff, F.; Kurjak, T.; Nouri, S.; Auer, M.K.; Wiedemann, K.; Briken, P.; Haaker, J.; Lonsdorf, T.B.; et al. An elevated plus-maze in mixed reality for studying human anxiety-related behavior. BMC Biol. 2017, 15, 125. [Google Scholar] [CrossRef]
- Arrant, A.E.; Schramm-Sapyta, N.L.; Kuhn, C.M. Use of the light/dark test for anxiety in adult and adolescent male rats. Behav. Brain Res. 2013, 256, 119–127. [Google Scholar] [CrossRef]
- Kwapis, J.L.; Alaghband, Y.; Kramár, E.A.; López, A.J.; Vogel Ciernia, A.; White, A.O.; Shu, G.; Rhee, D.; Michael, C.M.; Montellier, E.; et al. Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat. Commun. 2018, 9, 3323. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Malvaez, M.; Kramar, E.; Matheos, D.P.; Arrizon, A.; Cabrera, S.M.; Lynch, G.; Greene, R.W.; Wood, M.A. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology 2011, 36, 1545–1556. [Google Scholar] [CrossRef]
- Català-Solsona, J.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J. Nr4a2 Transcription Factor in Hippocampal Synaptic Plasticity, Memory and Cognitive Dysfunction: A Perspective Review. Front. Mol. Neurosci. 2021, 14, 786226. [Google Scholar] [CrossRef]
- Wang, J.; Wu, S.; Sun, Y.; Fang, Y.; Wu, R.; Lu, J.; Qing, Z.; Liang, X.; Wang, Z.; Zhang, W.; et al. Interaction of COMT and KIBRA modulates the association between hippocampal structure and episodic memory performance in healthy young adults. Behav. Brain Res. 2020, 384, 112550. [Google Scholar] [CrossRef]
- Norrholm, S.D.; Jovanovic, T.; Smith, A.K.; Binder, E.; Klengel, T.; Conneely, K.; Mercer, K.B.; Davis, J.S.; Kerley, K.; Winkler, J.; et al. Differential Genetic and Epigenetic Regulation of catechol-O-methyltransferase is Associated with Impaired Fear Inhibition in Posttraumatic Stress Disorder. Front. Behav. Neurosci. 2013, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.; Greene, J.; Russ, E.; Dutra, L.; Westen, D. A multidimensional meta-analysis of psychotherapy for PTSD. Am. J. Psychiatry 2005, 162, 214–227. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Name | Amplicon | Forward Sequence | Reverse Sequence |
|---|---|---|---|---|
| ADRB1 | Beta adrenoceptor 1 | 219 | ATCGTGCTGCTCATCGTAGT | TAGCACGTCTACCGAAGTCC |
| ADRB2 | Beta adrenoceptor 2 | 209 | GAGCACAAAGCCCTCAAGAC | TGGAAGGCAATCCTGAAATC |
| ADRA1D | Alpha adrenoceptor 1D | 135 | GGAAAAGATCCGTGGACAGT | AGCGGAAGAGCAACAGATTT |
| NR3C1 | nuclear receptor 3C1 | 192 | GCTTCAGGATGTCATTACGG | TCGAGCTTCAAGGTTCATTC |
| NR3C2 | nuclear receptor 3C2 | 109 | ACGCTGTGAGACTGGATTTC | AGTTACCCGGAGACACATGA |
| GRIA1 | AMPA receptor subunit 1 | 157 | CAAGGAACTGCAGGAAGAAA | CTAGAAAACCGGTGCAGAAA |
| GRIA2 | AMPA receptor subunit 2 | 109 | GAGGAAGAAAGGGAAACGAG | TCAGTCCCCATAAAACAGGA |
| GRIN2a | NMDA receptor subunit 2A | 278 | GCTGTCAGCACTGAATCCAA | GCCATTGACCGTTTGAAGTT |
| GRIN2b | NMDA receptor subunit 2B | 194 | CATCGTCACCACCTACTTCC | CCTTCGTGCAATAAAGGAGA |
| PER1 | Period circadian regulator 1 | 60 | AGAACAAGGTGGGAGCTCTT | TAGCTGGTGCCATTCTCTTC |
| EGR1 | Early growth response 1 | 100 | CGCTCACTCCACTATCCACT | GGTTTGATGAGTTGGGACTG |
| HDAC3 | Histone deacetylase 3 | 211 | CCCCCTTTCCTCAAACTCTC | TTGCATGGAAGCAAGAACTG |
| CREBBP | CREB binding protein | 60 | CAGAGACAAGCACTGGGAGT | ATGCACAGAGTGGACCATTT |
| COMT | Catechol-O-methyltransferase | 145 | AATGTCCAGACGCCAAATAA | CTGGATACTGGGGATGACAG |
| NR4A2 | Nuclear receptor 4A2 | 61 | TCTCCTGACTGGCTCTATGG | AGCAAAGCCAGGAATCTTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winzenried, E.T.; Everett, A.C.; Saito, E.R.; Miller, R.M.; Johnson, T.; Neal, E.; Boyce, Z.; Smith, C.; Jensen, C.; Kimball, S.; et al. Effects of a True Prophylactic Treatment on Hippocampal and Amygdala Synaptic Plasticity and Gene Expression in a Rodent Chronic Stress Model of Social Defeat. Int. J. Mol. Sci. 2023, 24, 11193. https://doi.org/10.3390/ijms241311193
Winzenried ET, Everett AC, Saito ER, Miller RM, Johnson T, Neal E, Boyce Z, Smith C, Jensen C, Kimball S, et al. Effects of a True Prophylactic Treatment on Hippocampal and Amygdala Synaptic Plasticity and Gene Expression in a Rodent Chronic Stress Model of Social Defeat. International Journal of Molecular Sciences. 2023; 24(13):11193. https://doi.org/10.3390/ijms241311193
Chicago/Turabian StyleWinzenried, Eric T., Anna C. Everett, Erin R. Saito, Roxanne M. Miller, Taylor Johnson, Eliza Neal, Zachary Boyce, Calvin Smith, Chloe Jensen, Spencer Kimball, and et al. 2023. "Effects of a True Prophylactic Treatment on Hippocampal and Amygdala Synaptic Plasticity and Gene Expression in a Rodent Chronic Stress Model of Social Defeat" International Journal of Molecular Sciences 24, no. 13: 11193. https://doi.org/10.3390/ijms241311193
APA StyleWinzenried, E. T., Everett, A. C., Saito, E. R., Miller, R. M., Johnson, T., Neal, E., Boyce, Z., Smith, C., Jensen, C., Kimball, S., Brantley, A., Melendez, G., Moffat, D., Davis, E., Aponik, L., Crofts, T., Dabney, B., & Edwards, J. G. (2023). Effects of a True Prophylactic Treatment on Hippocampal and Amygdala Synaptic Plasticity and Gene Expression in a Rodent Chronic Stress Model of Social Defeat. International Journal of Molecular Sciences, 24(13), 11193. https://doi.org/10.3390/ijms241311193