

Involvement of the p38/MK2 Pathway in MCLR Hepatotoxicity Revealed through MAPK Pharmacological Inhibition and Phosphoproteomics in HepaRG Cells

 ,
,
Abstract
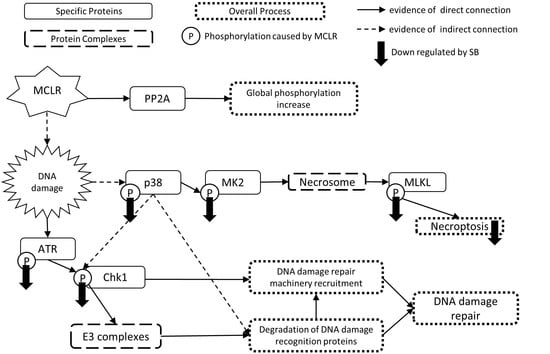
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Viability Assays
2.4. Immunoblot Assays
2.5. Phosphoproteomics
2.5.1. Protein Digestion and Phosphopeptide Enrichment
2.5.2. LC-MS Analysis
2.5.3. Phosphosite Analysis
2.6. Statistical Analysis
3. Results
3.1. MCLR-Elicited Viability Changes and Pharmacological Intervention in HepaRG Cells
3.2. Phosphoproteomics Analysis after MCLR Toxicity and/or p38 Inhibition
3.3. Mechanisms of p38 Inhibition for Rescue of MCLR Toxicity
3.3.1. Protein Phosphatase Expression, MCLR Protein Binding, and p38/MK2 Phosphorylation
3.3.2. Necroptosis, DNA Damage, and Protein Degradation Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Preece, E.P.; Hardy, F.J.; Moore, B.C.; Bryan, M. A review of microcystin detections in Estuarine and Marine waters: Environmental implications and human health risk. Harmful Algae 2017, 61, 31–45. [Google Scholar] [CrossRef]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Arman, T.; Clarke, J. Microcystin Toxicokinetics, Molecular Toxicology, and Pathophysiology in Preclinical Rodent Models and Humans. Toxins 2021, 13, 537. [Google Scholar] [CrossRef]
- Chen, L.; Giesy, J.P.; Adamovsky, O.; Svirčev, Z.; Meriluoto, J.; Codd, G.A.; Mijovic, B.; Shi, T.; Tuo, X.; Li, S.-C.; et al. Challenges of using blooms of Microcystis spp. in animal feeds: A comprehensive review of nutritional, toxicological and microbial health evaluation. Sci. Total Environ. 2021, 764, 142319. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zeng, H.; Lin, H.; Wang, J.; Feng, X.; Qiu, Z.; Chen, J.-A.; Luo, J.; Luo, Y.; Huang, Y.; et al. Serum microcystin levels positively linked with risk of hepatocellular carcinoma: A case-control study in southwest China. Hepatology 2017, 66, 1519–1528. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Agents Classified by the IARC Monographs, Volumes 1–132. 2022. Available online: https://www.iarc.who.int (accessed on 16 December 2022).
- World Health Organization. Cyanobacterial Toxins: Microcystins Background Document for Development of WHO. 2020. Available online: http://apps.who.int/bookorders (accessed on 16 December 2022).
- Komatsu, M.; Furukawa, T.; Ikeda, R.; Takumi, S.; Nong, Q.; Aoyama, K.; Akiyama, S.-I.; Keppler, D.; Takeuchi, T. Involvement of mitogen-activated protein kinase signaling pathways in microcystin-LR-induced apoptosis after its selective uptake mediated by OATP1B1 and OATP1B3. Toxicol. Sci. 2007, 97, 407–416. [Google Scholar] [CrossRef]
- Liu, J.; Sun, Y. The role of PP2A-associated proteins and signal pathways in microcystin-LR toxicity. Toxicol. Lett. 2015, 236, 1–7. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, J.; Xia, Z. Microcystin-LR Exhibits Immunomodulatory Role in Mouse Primary Hepatocytes through Activation of the NF-κB and MAPK Signaling Pathways. Toxicol. Sci. 2013, 136, 86–96. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, X.; Li, M.; Liu, J. P44/42 MAPK signal pathway-mediated hyperphosphorylation of paxillin and redistribution of E-cadherin was involved in microcystin-LR-reduced cellular adhesion in a human liver cell line. Chemosphere 2018, 200, 594–602. [Google Scholar] [CrossRef]
- Liu, J.; Wang, B.; Huang, P.; Wang, H.; Xu, K.; Wang, X.; Xu, L.; Guo, Z. Microcystin-LR promotes cell proliferation in the mice liver by activating Akt and p38/ERK/JNK cascades. Chemosphere 2016, 163, 14–21. [Google Scholar] [CrossRef]
- Marion, M.J.; Hantz, O.; Durantel, D. The HepaRG cell line: Biological properties and relevance as a tool for cell biology, drug metabolism, and virology studies. Methods Mol. Biol. 2010, 640, 261–272. [Google Scholar] [PubMed]
- Hart, S.; Li, Y.; Nakamoto, K.; Subileau, E.-A.; Steen, D.; Zhong, X.-B. A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues. Drug Metab. Dispos. 2010, 38, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Kotani, N.; Maeda, K.; Debori, Y.; Camus, S.; Li, R.; Chesne, C.; Sugiyama, Y. Expression and transport function of drug uptake transporters in differentiated HepaRG cells. Mol. Pharm. 2012, 9, 3434–3441. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Zhou, Y.-S.; Yang, J.-X.; Zhao, Y.-L.; Liu, Q.-Q.; Yuan, C.; Wang, F.-Z. MK-2206 induces cell cycle arrest and apoptosis in HepG2 cells and sensitizes TRAIL-mediated cell death. Mol. Cell. Biochem. 2013, 382, 217–224. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681. [Google Scholar] [CrossRef]
- Xue, L.; Li, J.; Li, Y.; Chu, C.; Xie, G.; Qin, J.; Yang, M.; Zhuang, D.; Cui, L.; Zhang, H.; et al. N-acetylcysteine protects Chinese Hamster ovary cells from oxidative injury and apoptosis induced by microcystin-LR. Int. J. Clin. Exp. Med. 2015, 8, 4911–4921. [Google Scholar]
- Aldridge, G.M.; Podrebarac, D.M.; Greenough, W.T.; Weiler, I.J. The use of total protein stains as loading controls: An alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 2008, 172, 250–254. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, T.; Xiao, D.; Yang, P. Protocol for the processing and downstream analysis of phosphoproteomic data with PhosR. STAR Protoc. 2021, 2, 100585. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, T.; Hoffman, N.J.; Xiao, D.; James, D.E.; Humphrey, S.J.; Yang, P. PhosR enables processing and functional analysis of phosphoproteomic data. Cell Rep. 2021, 34, 108771. [Google Scholar] [CrossRef]
- Mikhailov, A.; Härmälä-Braskén, A.S.; Hellman, J.; Meriluoto, J.; Eriksson, J.E. Identification of ATP-synthase as a novel intracellular target for microcystin-LR. Chem.-Biol. Interact. 2003, 142, 223–237. [Google Scholar] [CrossRef]
- Runnegar, M.; Berndt, N.; Kong, S.M.; Lee, E.Y.C.; Zhang, L.F. In Vivo and in Vitro Binding of Microcystin to Protein Phosphatase 1 and 2A. Biochem. Biophys. Res. Commun. 1995, 216, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef]
- Bonney, E.A. Mapping out p38MAPK. Am. J. Reprod. Immunol. 2017, 77, e12652. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wu, J.; Silke, J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Res 2020, 9, F1000, Faculty Rev-653. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Meng, G.; Guo, Z.; Xu, L. Regulation of heat shock protein 27 phosphorylation during microcystin-LR-induced cytoskeletal reorganization in a human liver cell line. Toxicol. Lett. 2011, 207, 270–277. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, J.; Huang, P.; Guo, Z.; Xu, L. Alterations of tau and VASP during microcystin-LR-induced cytoskeletal reorganization in a human liver cell line. Environ. Toxicol. 2015, 30, 92–100. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, T.; Lei, T.; Zhang, D.; Du, S.; Girani, L.; Qi, D.; Lin, C.; Tong, R.; Wang, Y. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int. J. Mol. Med. 2019, 44, 771–786. [Google Scholar] [CrossRef]
- Morgan, M.J.; Kim, Y.-S. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 2022, 54, 1695–1704. [Google Scholar] [CrossRef]
- Seifert, L.; Miller, G. Molecular Pathways: The Necrosome-A Target for Cancer Therapy. Clin. Cancer Res. 2017, 23, 1132–1136. [Google Scholar] [CrossRef]
- Wu, Y.-L.; He, Y.; Shi, J.-J.; Zheng, T.-X.; Lin, X.-J.; Lin, X. Microcystin-LR promotes necroptosis in primary mouse hepatocytes by overproducing reactive oxygen species. Toxicol. Appl. Pharmacol. 2019, 377, 114626. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xie, P. Mechanisms of Microcystin-induced Cytotoxicity and Apoptosis. Mini-Rev. Med. Chem. 2016, 16, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Weng, D.; Li, F.; Zou, X.; Young, D.O.; Ji, J.; Shen, P. Involvement of JNK regulation in oxidative stress-mediated murine liver injury by microcystin-LR. Apoptosis 2008, 13, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Matsushima-Nishiwaki, R.; Shidoji, Y.; Nishiwaki, S.; Yamada, T.; Moriwaki, H.; Muto, Y. Suppression by carotenoids of microcystin-induced morphological changes in mouse hepatocytes. Lipids 1995, 30, 1029–1034. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Xiao, H.; Zhang, M.; Bao, T.; Luo, X.; Chen, S. Genotoxicity of microcystin-LR in mammalian cells: Implication from peroxynitrite produced by mitochondria. Ecotoxicol. Environ. Saf. 2020, 195, 110408. [Google Scholar] [CrossRef] [PubMed]
- Žegura, B.; Sedmak, B.; Filipič, M. Microcystin-LR induces oxidative DNA damage in human hepatoma cell line HepG2. Toxicon 2003, 41, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Sakamoto, H.; Sakuraba, M.; Wu, D.S.; Zhang, L.S.; Suzuki, T.; Hayashi, M.; Honma, M. Genotoxicity of microcystin-LR in human lymphoblastoid TK6 cells. Mutat. Res. 2004, 557, 1–6. [Google Scholar] [CrossRef]
- Žegura, B.; Gajski, G.; Štraser, A.; Garaj-Vrhovac, V.; Filipič, M. Microcystin-LR induced DNA damage in human peripheral blood lymphocytes. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2011, 726, 116–122. [Google Scholar] [CrossRef]
- Liu, W.; Wang, L.; Zheng, C.; Liu, L.; Wang, J.; Li, D.; Tan, Y.; Zhao, X.; He, L.; Shu, W. Microcystin-LR increases genotoxicity induced by aflatoxin B1 through oxidative stress and DNA base excision repair genes in human hepatic cell lines. Environ. Pollut. 2018, 233, 455–463. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Poon, R.Y.C. Aurora kinases and DNA damage response. Mutat. Res. 2020, 821, 111716. [Google Scholar] [CrossRef] [PubMed]
- Krupina, K.; Goginashvili, A.; Cleveland, D.W. Causes and consequences of micronuclei. Curr. Opin. Cell Biol. 2021, 70, 91–99. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, H.; Chen, X.; Losiewicz, M.D.; Wang, R.; Du, X.; Wang, B.; Ma, Y.; Zhang, S.; Shi, L.; et al. The activated ATM/p53 pathway promotes autophagy in response to oxidative stress-mediated DNA damage induced by Microcystin-LR in male germ cells. Ecotoxicol. Environ. Saf. 2021, 227, 112919. [Google Scholar] [CrossRef]
- Bao, N.; Han, J.; Zhou, H. A protein with broad functions: Damage-specific DNA-binding protein 2. Mol. Biol. Rep. 2022, 49, 12181–12192. [Google Scholar] [CrossRef]
- Sugasawa, K.; Okuda, Y.; Saijo, M.; Nishi, R.; Matsuda, N.; Chu, G.; Mori, T.; Iwai, S.; Tanaka, K.; Tanaka, K.; et al. UV-Induced Ubiquitylation of XPC Protein Mediated by UV-DDB-Ubiquitin Ligase Complex. Cell 2005, 121, 387–400. [Google Scholar] [CrossRef]
- Roy, N.; Elangovan, I.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. Tumor regression by phenethyl isothiocyanate involves DDB2. Cancer Biol. Ther. 2013, 14, 108–116. [Google Scholar] [CrossRef]
- Matsumoto, S.; Fischer, E.; Yasuda, T.; Dohmae, N.; Iwai, S.; Mori, T.; Nishi, R.; Yoshino, K.-i.; Sakai, W.; Hanaoka, F.; et al. Functional regulation of the DNA damage-recognition factor DDB2 by ubiquitination and interaction with xeroderma pigmentosum group C protein. Nucleic Acids Res. 2015, 43, 1700–1713. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCLR Reactome | p-Value | Number of Substrates | SB203580 Reactome | p-Value | Number of Substrates |
|---|---|---|---|---|---|
| Activation of anterior HOX genes in hindbrain development during early embryogenesis | 0.020 | 9 | Activation of anterior HOX genes in hindbrain development during early embryogenesis | 0.029 | 9 |
| Activation of HOX genes during differentiation | 0.020 | 9 | Activation of HOX genes during differentiation | 0.029 | 9 |
| Apoptosis | 0.038 | 34 | Activation of the AP-1 family of transcription factors | 0.043 | 5 |
| Apoptotic cleavage of cellular proteins | 0.035 | 16 | Asparagine N-linked glycosylation | 0.026 | 35 |
| Apoptotic execution phase | 0.011 | 18 | Aspirin ADME | 0.043 | 4 |
| Carnitine metabolism | 0.045 | 5 | Chromosome maintenance | 0.045 | 11 |
| Cell junction organization | 0.027 | 19 | COPI-dependent Golgi-to-ER retrograde traffic | 0.024 | 16 |
| Condensation of prophase chromosomes | 0.019 | 4 | Diseases of glycosylation | 0.032 | 5 |
| COPII-mediated vesicle transport | 0.049 | 12 | DNA damage bypass | 0.018 | 8 |
| Cytoprotection by HMOX1 | 0.049 | 12 | ER-to-Golgi anterograde transport | 0.047 | 28 |
| Depolymerization of the nuclear lamina | 0.015 | 9 | Gap junction trafficking | 0.037 | 4 |
| Disassembly of the destruction complex and recruitment of AXIN to the membrane | 0.005 | 4 | Gap junction trafficking and regulation | 0.012 | 5 |
| ERK/MAPK targets | 0.019 | 6 | Gluconeogenesis | 0.000 | 7 |
| Estrogen-dependent gene expression | 0.048 | 18 | Glycogen synthesis | 0.012 | 4 |
| Gene and protein expression by JAK-STAT signaling after interleukin-12 stimulation | 0.043 | 8 | Hemostasis | 0.007 | 73 |
| Glycogen breakdown (glycogenolysis) | 0.031 | 5 | Kinesins | 0.039 | 9 |
| Glycogen metabolism | 0.045 | 7 | L1CAM interactions | 0.017 | 20 |
| Initiation of nuclear envelope (NE) reformation | 0.023 | 8 | MAPK targets/nuclear events mediated by MAP kinases | 0.009 | 8 |
| Inositol phosphate metabolism | 0.008 | 4 | Membrane trafficking | 0.026 | 123 |
| Interleukin-12 family signaling | 0.016 | 12 | Metabolism of carbohydrates | 0.022 | 38 |
| Interleukin-12 signaling | 0.043 | 8 | Mitochondrial biogenesis | 0.048 | 16 |
| IRE1alpha activates chaperones | 0.028 | 10 | NOTCH3 activation and transmission of signal to the nucleus | 0.046 | 5 |
| Meiosis | 0.035 | 8 | Nuclear Events (kinase and transcription factor activation) | 0.019 | 13 |
| Meiotic synapsis | 0.035 | 8 | Peptide hormone metabolism | 0.047 | 4 |
| Metabolism of carbohydrates | 0.010 | 38 | Platelet activation, signaling, and aggregation | 0.032 | 34 |
| Muscle contraction | 0.002 | 20 | Platelet degranulation | 0.002 | 17 |
| Platelet degranulation | 0.038 | 17 | Post-translational protein phosphorylation | 0.044 | 13 |
| Platelet homeostasis | 0.040 | 10 | Regulation of beta-cell development | 0.021 | 4 |
| PPARA activates gene expression | 0.009 | 18 | Regulation of insulin-like growth factor (IGF) transport and uptake by insulin-like growth factor binding proteins (IGFBPs) | 0.044 | 13 |
| Recycling pathway of L1 | 0.007 | 9 | Regulation of TP53 degradation | 0.013 | 8 |
| Regulation of glycolysis by fructose 2,6-bisphosphate metabolism | 0.026 | 4 | Regulation of TP53 expression and degradation | 0.013 | 8 |
| Regulation of lipid metabolism by PPARalpha | 0.005 | 19 | Response to elevated platelet cytosolic Ca2+ | 0.003 | 18 |
| Regulation of TP53 activity through acetylation | 0.021 | 7 | RHOU GTPase cycle | 0.045 | 17 |
| Reproduction | 0.035 | 8 | Signaling by ERBB4 | 0.036 | 8 |
| Response to elevated platelet cytosolic Ca2+ | 0.026 | 18 | Signaling by NTRK1 (TRKA) | 0.029 | 25 |
| RHO GTPases activate IQGAPs | 0.033 | 7 | Signaling by NTRKs | 0.016 | 27 |
| RHOD GTPase cycle | 0.004 | 19 | Termination of translesion DNA synthesis | 0.027 | 4 |
| RHOF GTPase cycle | 0.006 | 16 | Toll-like receptor 4 (TLR4) cascade | 0.043 | 16 |
| RORA activates gene expression | 0.046 | 5 | TP53 regulates transcription of several additional cell death genes whose specific roles in p53-dependent apoptosis remain uncertain | 0.036 | 4 |
| SARS-CoV-2 activates/modulates innate and adaptive immune responses | 0.034 | 22 | Transport to the Golgi and subsequent modification | 0.047 | 28 |
| Signaling by NOTCH1 | 0.046 | 11 | Vesicle-mediated transport | 0.025 | 127 |
| Smooth muscle contraction | 0.015 | 11 | |||
| Sphingolipid metabolism | 0.030 | 8 | |||
| Synthesis of bile acids and bile salts via 7alpha-hydroxycholesterol | 0.029 | 4 | |||
| Transcriptional regulation of white adipocyte differentiation | 0.032 | 13 | |||
| Transport of small molecules | 0.025 | 63 | |||
| Unfolded protein response (UPR) | 0.019 | 14 | |||
| XBP1(S) activates chaperone genes | 0.028 | 10 |
| Upregulated | Downregulated | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Site | LogFC | A.p.Val | Gene | Site | LogFC | A.P.Val | Gene | Site | LogFC | A.P.Val | Gene | Site | LogFC | A.P.Val | Gene | Site | LogFC | A.P.Val |
| AAK1 | 456 | 1.50 | 0.023 | ANKS6 | 20 | −1.00 | 0.033 | EPB41L1 | 510 | −1.24 | 0.044 | LOC113230 | 233 | −1.53 | 0.006 | RBM15 | 612 | −1.19 | 0.014 |
| AAK1 | 249 | 1.14 | 0.023 | BAG6 | 994 | −2.77 | 0.004 | EPB41L1 | 783 | −1.07 | 0.045 | MAP4K4 | 981 | −0.97 | 0.046 | RBM15 | 656 | −1.00 | 0.014 |
| BET1L | 9 | 0.96 | 0.038 | BAG6 | 1003 | −1.53 | 0.006 | F11R | 264 | −1.28 | 0.016 | MTSS1L | 455 | −1.56 | 0.016 | RPUSD2 | 35 | −1.26 | 0.016 |
| CDC42BPA | 735 | 1.01 | 0.031 | BSG | 166 | −1.57 | 0.008 | FAM21C | 245 | −1.41 | 0.025 | NA | 308 | −0.79 | 0.049 | SCRIB | 1486 | −1.29 | 0.046 |
| CDK11B | 234 | 0.71 | 0.044 | CLN6 | 7 | −0.96 | 0.049 | GPALPP1 | 103 | −1.25 | 0.029 | NKTR | 79 | −1.53 | 0.017 | SCRIB | 1475 | −1.05 | 0.045 |
| CLASRP | 285 | 1.41 | 0.025 | CUL4A | 10 | −1.32 | 0.014 | GPRC5C | 29 | −0.98 | 0.036 | NOLC1 | 707 | −0.92 | 0.033 | SCRIB | 1378 | −1.58 | 0.046 |
| CTDP1 | 672 | 1.14 | 0.025 | DDB2 | 26 | −1.39 | 0.009 | HDLBP | 31 | −2.31 | 0.0002 | PARD3B | 924 | −1.52 | 0.019 | SLC4A1AP | 258 | −1.09 | 0.019 |
| DDX52 | 22 | 1.69 | 0.007 | DDB2 | 24 | −1.41 | 0.013 | HNRNPM | 481 | −1.36 | 0.019 | PHRF1 | 1202 | −1.23 | 0.046 | SMARCA2 | 1377 | −0.77 | 0.047 |
| ELMSAN1 | 461 | 2.09 | 0.035 | DDX46 | 804 | −0.95 | 0.036 | HNRNPM | 575 | −1.57 | 0.023 | PHRF1 | 936 | −1.10 | 0.047 | SPAG9 | 203 | −1.05 | 0.023 |
| EPB41L3 | 448 | 1.16 | 0.028 | DLG1 | 586 | −0.98 | 0.035 | HNRNPM | 452 | −2.09 | 0.023 | PHRF1 | 867 | −1.10 | 0.046 | SPTAN1 | 1197 | −0.86 | 0.045 |
| GPRC5C | 68 | 0.98 | 0.035 | EEF1D | 162 | −1.06 | 0.023 | HNRNPM | 513 | −1.00 | 0.023 | PHRF1 | 991 | −1.27 | 0.049 | SYNE1 | 7900 | −1.21 | 0.037 |
| HNRNPM | 528 | 1.04 | 0.022 | EFR3A | 721 | −1.06 | 0.049 | HNRNPM | 432 | −1.00 | 0.023 | PIK3C2A | 338 | −0.87 | 0.038 | USP36 | 742 | −2.13 | 0.007 |
| SLC26A6 | 644 | 2.04 | 0.036 | EIF4G3 | 494 | −0.92 | 0.033 | HNRNPM | 86 | −1.20 | 0.023 | PIK3C2A | 259 | −0.91 | 0.038 | VGLL4 | 59 | −1.53 | 0.006 |
| SRCIN1 | 1021 | 1.39 | 0.023 | EIF5B | 113 | −1.83 | 0.008 | HNRNPUL1 | 94 | −1.20 | 0.029 | PKP4 | 314 | −1.29 | 0.046 | VPS11 | 803 | −1.79 | 0.016 |
| ZMYND8 | 695 | 1.27 | 0.014 | EIF5B | 164 | −1.53 | 0.008 | HSPH1 | 73 | −1.07 | 0.033 | PPAN | 306 | −0.82 | 0.035 | ZCCHC17 | 52 | −2.20 | 0.016 |
| ZNF446 | 3 | 1.57 | 0.007 | EIF5B | 214 | −1.52 | 0.008 | IQSEC1 | 166 | −1.17 | 0.014 | RAB11FIP5 | 307 | −1.13 | 0.047 | ZSCAN29 | 153 | −1.13 | 0.037 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynch, K.D.; Iverson, D.T.; Bachhav, N.K.; Call, M.R.; Yue, G.E.; Prasad, B.; Clarke, J.D. Involvement of the p38/MK2 Pathway in MCLR Hepatotoxicity Revealed through MAPK Pharmacological Inhibition and Phosphoproteomics in HepaRG Cells. Int. J. Mol. Sci. 2023, 24, 11168. https://doi.org/10.3390/ijms241311168
Lynch KD, Iverson DT, Bachhav NK, Call MR, Yue GE, Prasad B, Clarke JD. Involvement of the p38/MK2 Pathway in MCLR Hepatotoxicity Revealed through MAPK Pharmacological Inhibition and Phosphoproteomics in HepaRG Cells. International Journal of Molecular Sciences. 2023; 24(13):11168. https://doi.org/10.3390/ijms241311168
Chicago/Turabian StyleLynch, Katherine D., Dayne T. Iverson, Namrata K. Bachhav, Michael Ridge Call, Guihua Eileen Yue, Bhagwat Prasad, and John D. Clarke. 2023. "Involvement of the p38/MK2 Pathway in MCLR Hepatotoxicity Revealed through MAPK Pharmacological Inhibition and Phosphoproteomics in HepaRG Cells" International Journal of Molecular Sciences 24, no. 13: 11168. https://doi.org/10.3390/ijms241311168
APA StyleLynch, K. D., Iverson, D. T., Bachhav, N. K., Call, M. R., Yue, G. E., Prasad, B., & Clarke, J. D. (2023). Involvement of the p38/MK2 Pathway in MCLR Hepatotoxicity Revealed through MAPK Pharmacological Inhibition and Phosphoproteomics in HepaRG Cells. International Journal of Molecular Sciences, 24(13), 11168. https://doi.org/10.3390/ijms241311168