
MCL1 Inhibition Overcomes the Aggressiveness Features of Triple-Negative Breast Cancer MDA-MB-231 Cells

 , ,
, ,  ,
,  ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effects of A-1210477 on MDA-MD-231 Cell Viability and Morphology
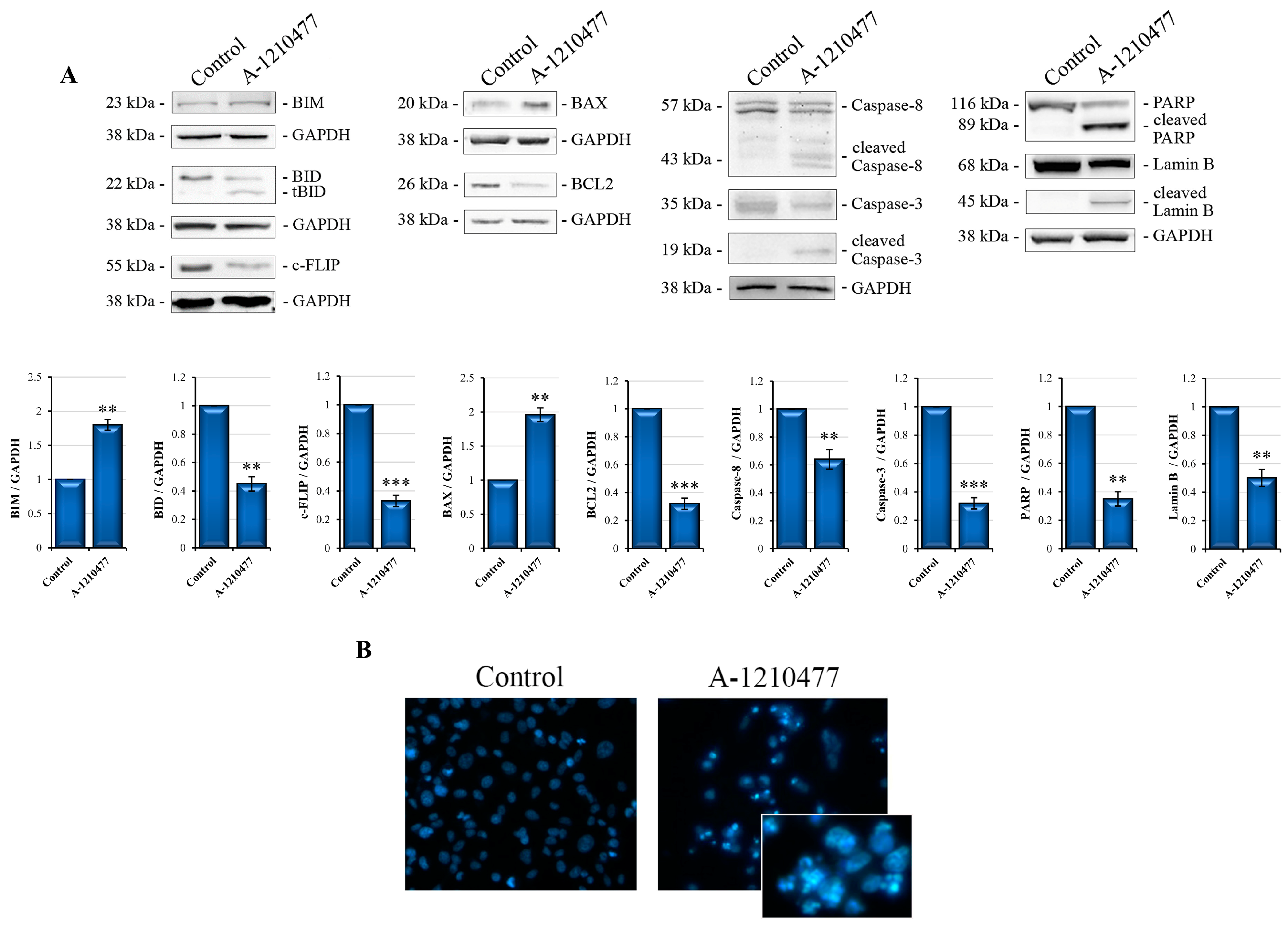
2.2. A-1210477 Treatment Induces Anoikis in MDA-MB-231 Cells
2.3. MCL1 Inhibition Promotes Integrin Switch along with Focal Adhesions (FAs) and Survival Pathway Interruption
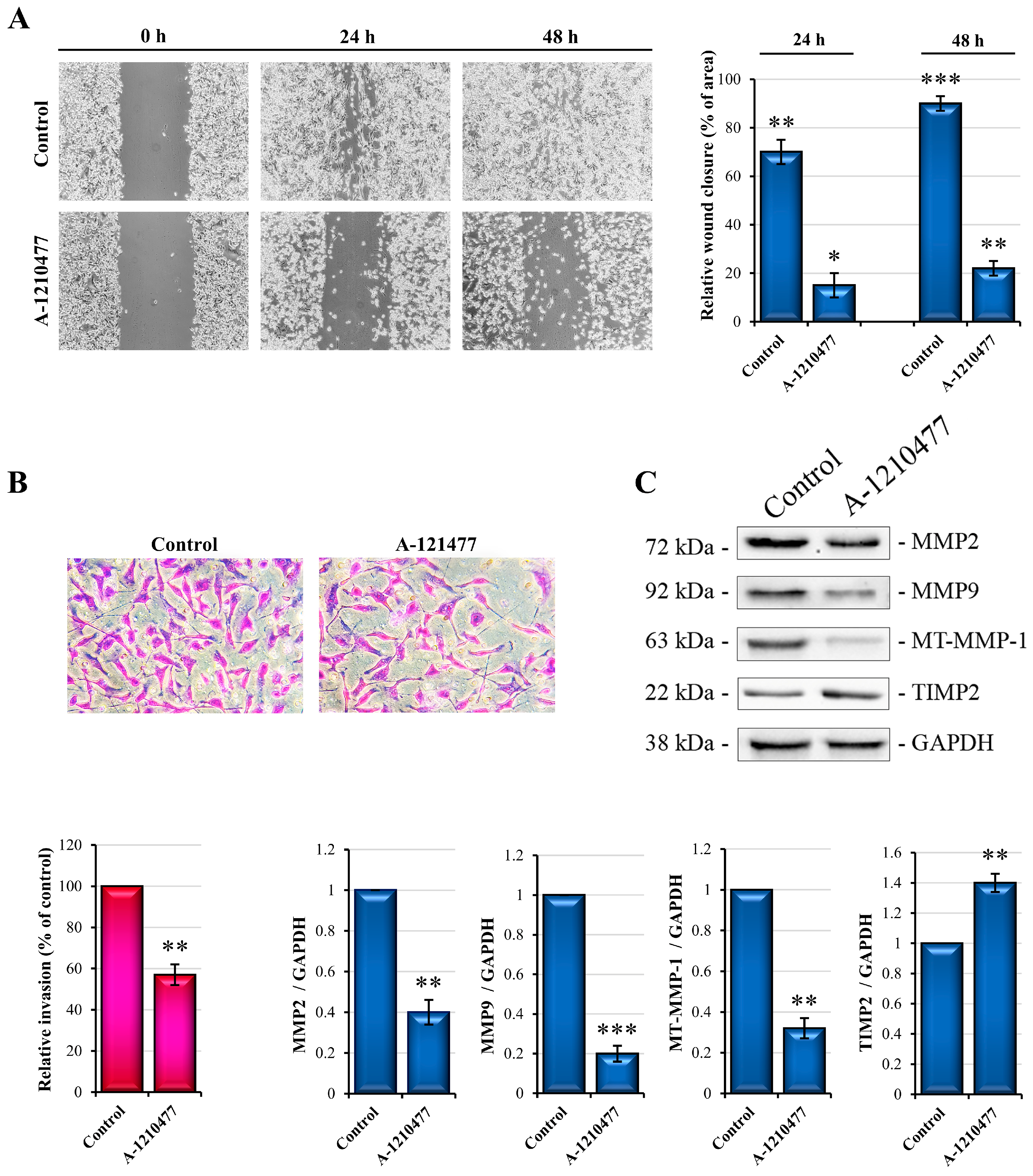
2.4. MCL1 Inhibition Reduces Migration and Invasiveness in MDA-MB-231 Cells
2.5. MCL1 Inhibition Reduces Stemness Features in MDA-MB-231 Cells
2.6. MCL1 Inhibition Promotes a Possible Reversion of Epithelial–Mesenchymal Transition (EMT)
2.7. The De Novo Production of E-Cadherin via GSK3β Activity and DNMT1 Degradation
2.8. MCL1 Inhibition Promotes Itself Downregulation
2.9. A Possible Non-Canonical Localization and Role of MCL1 in FAs
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions and Reagents
4.2. Cell Viability Assay
4.3. Western Blot and Immunoprecipitation Analysis
4.4. Wound Healing Assay
4.5. Transwell Migration Assay
4.6. Flow Cytometry Analysis of Cell Surface Marker Expression
4.7. qRT-PCR Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef]
- Siddig, A.; Tengku Din, T.A.D.A.; Mohd Nafi, S.N.; Yahya, M.M.; Sulong, S.; Wan Abdul Rahman, W.F. The Unique Biology behind the Early Onset of Breast Cancer. Genes 2021, 12, 372. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qin, S.; Yi, Y.; Gao, H.; Liu, X.; Ma, F.; Guan, M. Delving into the Heterogeneity of Different Breast Cancer Subtypes and the Prognostic Models Utilizing scRNA-Seq and Bulk RNA-Seq. Int. J. Mol. Sci. 2022, 23, 9936. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Pal, D.; Sharma, R.; Garg, V.K.; Goel, N.; Koundal, D.; Zaguia, A.; Koundal, S.; Belay, A. Global Increase in Breast Cancer Incidence: Risk Factors and Preventive Measures. Biomed Res. Int. 2022, 2022, 9605439. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Ghorayeb, T.; Saliba, F.; Allam, S.; Bou Zerdan, M.; Yaghi, M.; Bilani, N.; Jaafar, R.; Nahleh, Z. Triple Negative Breast Cancer: Updates on Classification and Treatment in 2021. Cancers 2022, 14, 1253. [Google Scholar] [CrossRef]
- So, J.Y.; Ohm, J.; Lipkowitz, S.; Yang, L. Triple negative breast cancer (TNBC): Non-genetic tumor heterogeneity and immune microenvironment: Emerging treatment options. Pharmacol. Ther. 2022, 237, 108253. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef]
- de Sousa Mesquita, A.P.; de Araújo Lopes, S.; Pernambuco Filho, P.C.A.; Nader, H.B.; Lopes, C.C. Acquisition of anoikis resistance promotes alterations in the Ras/ERK and PI3K/Akt signaling pathways and matrix remodeling in endothelial cells. Apoptosis 2017, 22, 1116–1137. [Google Scholar] [CrossRef]
- Fanfone, D.; Wu, Z.; Mammi, J.; Berthenet, K.; Neves, D.; Weber, K.; Halaburkova, A.; Virard, F.; Bunel, F.; Jamard, C.; et al. Confined migration promotes cancer metastasis through resistance to anoikis and increased invasiveness. eLife 2022, 11, e73150. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Kang, Y. Stresses in the metastatic cascade: Molecular mechanisms and therapeutic opportunities. Genes Dev. 2020, 34, 1577–1598. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rivandi, M.; Abedini, S.; Pasdar, A.; Sahebkar, A. Regulators and mechanisms of anoikis in triple-negative breast cancer (TNBC): A review. Crit. Rev. Oncol. Hematol. 2019, 140, 17–27. [Google Scholar] [CrossRef]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Kvokačková, B.; Remšík, J.; Jolly, M.K.; Souček, K. Phenotypic Heterogeneity of Triple-Negative Breast Cancer Mediated by Epithelial-Mesenchymal Plasticity. Cancers 2021, 13, 2188. [Google Scholar] [CrossRef]
- Xue, W.; Sun, C.; Yuan, H.; Yang, X.; Zhang, Q.; Liao, Y.; Guo, H. Establishment and Analysis of an Individualized EMT-Related Gene Signature for the Prognosis of Breast Cancer in Female Patients. Dis. Markers 2022, 2022, 1289445. [Google Scholar] [CrossRef] [PubMed]
- Khaled, N.; Bidet, Y. New Insights into the Implication of Epigenetic Alterations in the EMT of Triple Negative Breast Cancer. Cancers 2019, 11, 559. [Google Scholar] [CrossRef] [PubMed]
- Zolota, V.; Tzelepi, V.; Piperigkou, Z.; Kourea, H.; Papakonstantinou, E.; Argentou, Μ.I.; Karamanos, N.K. Epigenetic Alterations in Triple-Negative Breast Cancer-The Critical Role of Extracellular Matrix. Cancers 2021, 13, 713. [Google Scholar] [CrossRef]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef] [PubMed]
- Poddar, A.; Rao, S.R.; Prithviraj, P.; Kannourakis, G.; Jayachandran, A. Crosstalk between Immune Checkpoint Modulators, Metabolic Reprogramming and Cellular Plasticity in Triple-Negative Breast Cancer. Curr. Oncol. 2022, 29, 6847–6863. [Google Scholar] [CrossRef] [PubMed]
- Matadamas-Guzman, M.; Zazueta, C.; Rojas, E.; Resendis-Antonio, O. Analysis of Epithelial-Mesenchymal Transition Metabolism Identifies Possible Cancer Biomarkers Useful in Diverse Genetic Backgrounds. Front. Oncol. 2020, 10, 1309. [Google Scholar] [CrossRef]
- Fultang, N.; Chakraborty, M.; Peethambaran, B. Regulation of cancer stem cells in triple negative breast cancer. Cancer Drug Resist. 2021, 4, 321–342. [Google Scholar] [CrossRef]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Yang, W.T.; Rosen, D.G.; Landis, M.D.; Wong, H.; Lewis, M.T.; Creighton, C.J.; Sexton, K.R.; Hilsenbeck, S.G.; Sahin, A.A.; et al. Cancer stem cell markers are enriched in normal tissue adjacent to triple negative breast cancer and inversely correlated with DNA repair deficiency. Breast Cancer Res. 2013, 15, R77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef]
- Lisencu, L.A.; Trancă, S.; Bonci, E.A.; Pașca, A.; Mihu, C.; Irimie, A.; Tudoran, O.; Balacescu, O.; Lisencu, I.C. The Role of Circulating Tumor Cells in the Prognosis of Metastatic Triple-Negative Breast Cancers: A Systematic Review of the Literature. Biomedicines 2022, 10, 769. [Google Scholar] [CrossRef]
- De Blasio, A.; Vento, R.; Di Fiore, R. Mcl-1 targeting could be an intriguing perspective to cure cancer. J. Cell. Physiol. 2018, 233, 8482–8498. [Google Scholar] [CrossRef]
- Widden, H.; Placzek, W.J. The multiple mechanisms of MCL1 in the regulation of cell fate. Commun. Biol. 2021, 4, 1029. [Google Scholar] [CrossRef]
- Winder, M.L.; Campbell, K.J. MCL-1 is a clinically targetable vulnerability in breast cancer. Cell Cycle 2022, 21, 1439–1455. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Leiva, D.; Lucendo, E.; Orzáez, M. Understanding MCL1: From cellular function and regulation to pharmacological inhibition. FEBS J. 2022, 289, 6209–6234. [Google Scholar] [CrossRef] [PubMed]
- Young, A.I.; Timpson, P.; Gallego-Ortega, D.; Ormandy, C.J.; Oakes, S.R. Myeloid cell leukemia 1 (MCL-1), an unexpected modulator of protein kinase signaling during invasion. Cell Adh. Migr. 2018, 12, 513–523. [Google Scholar] [CrossRef]
- Carlisi, D.; D’Anneo, A.; Martinez, R.; Emanuele, S.; Buttitta, G.; Di Fiore, R.; Vento, R.; Tesoriere, G.; Lauricella, M. The oxygen radicals involved in the toxicity induced by parthenolide in MDA-MB-231 cells. Oncol. Rep. 2014, 32, 167–172. [Google Scholar] [CrossRef]
- Drago-Ferrante, R.; Pentimalli, F.; Carlisi, D.; De Blasio, A.; Saliba, C.; Baldacchino, S.; Degaetano, J.; Debono, J.; Caruana-Dingli, G.; Grech, G.; et al. Suppressive role exerted by microRNA-29b-1-5p in triple negative breast cancer through SPIN1 regulation. Oncotarget 2017, 8, 28939–28958. [Google Scholar] [CrossRef] [PubMed]
- De Blasio, A.; Di Fiore, R.; Pratelli, G.; Drago-Ferrante, R.; Saliba, C.; Baldacchino, S.; Grech, G.; Scerri, C.; Vento, R.; Tesoriere, G. A loop involving NRF2, miR-29b-1-5p and AKT, regulates cell fate of MDA-MB-231 triple-negative breast cancer cells. J. Cell. Physiol. 2020, 235, 629–637. [Google Scholar] [CrossRef]
- Lauricella, M.; Carlisi, D.; Giuliano, M.; Calvaruso, G.; Cernigliaro, C.; Vento, R.; D’Anneo, A. The analysis of estrogen receptor-α positive breast cancer stem-like cells unveils a high expression of the serpin proteinase inhibitor PI-9: Possible regulatory mechanisms. Int. J. Oncol. 2016, 49, 352–360. [Google Scholar] [CrossRef]
- De Blasio, A.; Pratelli, G.; Drago-Ferrante, R.; Saliba, C.; Baldacchino, S.; Grech, G.; Tesoriere, G.; Scerri, C.; Vento, R.; Di Fiore, R. Loss of MCL1 function sensitizes the MDA-MB-231 breast cancer cells to rh-TRAIL by increasing DR4 levels. J. Cell. Physiol. 2019, 234, 18432–18447. [Google Scholar] [CrossRef]
- Senichkin, V.V.; Pervushin, N.V.; Zamaraev, A.V.; Sazonova, E.V.; Zuev, A.P.; Streletskaia, A.Y.; Prikazchikova, T.A.; Zatsepin, T.S.; Kovaleva, O.V.; Tchevkina, E.M.; et al. Bak and Bcl-xL Participate in Regulating Sensitivity of Solid Tumor Derived Cell Lines to Mcl-1 Inhibitors. Cancers 2021, 14, 181. [Google Scholar] [CrossRef]
- Huang, Z.; Yu, P.; Tang, J. Characterization of Triple-Negative Breast Cancer MDA-MB-231 Cell Spheroid Model. Onco. Targets Ther. 2020, 13, 5395–5405. [Google Scholar] [CrossRef]
- Soutar, M.P.M.; Kempthorne, L.; Annuario, E.; Luft, C.; Wray, S.; Ketteler, R.; Ludtmann, M.H.R.; Plun-Favreau, H. FBS/BSA media concentration determines CCCP’s ability to depolarize mitochondria and activate PINK1-PRKN mitophagy. Autophagy 2019, 15, 2002–2011. [Google Scholar] [CrossRef]
- Sattari Fard, F.; Jalilzadeh, N.; Mehdizadeh, A.; Sajjadian, F.; Velaei, K. Understanding and targeting anoikis in metastasis for cancer therapies. Cell Biol. Int. 2023, 47, 683–698. [Google Scholar] [CrossRef]
- Giuliano, M.; Bellavia, G.; Lauricella, M.; D’Anneo, A.; Vassallo, B.; Vento, R.; Tesoriere, G. Staurosporine-induced apoptosis in Chang liver cells is associated with down-regulation of Bcl-2 and Bcl-XL. Int. J. Mol. Med. 2004, 13, 565–571. [Google Scholar] [CrossRef]
- Wolf, P.; Schoeniger, A.; Edlich, F. Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial membrane. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119317. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, B.; Shi, J.; Li, J.; Lu, X.; Xu, L.; Yang, H.; Hamad, N.; Wang, C.; Napier, D.; et al. BRD4 modulates vulnerability of triple-negative breast cancer to targeting of integrin-dependent signaling pathways. Cell Oncol. 2020, 43, 1049–1066. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Zavarella, S.; Symons, M.H. Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol. Cell. Biol. 2008, 28, 4162–4172. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, Z.; Luo, T.; Shi, H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol. Biomed. 2022, 3, 47. [Google Scholar] [CrossRef]
- Dofara, S.G.; Chang, S.L.; Diorio, C. Gene Polymorphisms and Circulating Levels of MMP-2 and MMP-9: A Review of Their Role in Breast Cancer Risk. Anticancer Res. 2020, 40, 3619–3631. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Morad, G.; Jedinak, A.; Moses, M.A. Metalloproteinases and their roles in human cancer. Anat. Rec. 2020, 303, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
- Hero, T.; Bühler, H.; Kouam, P.N.; Priesch-Grzeszowiak, B.; Lateit, T.; Adamietz, I.A. The Triple-negative Breast Cancer Cell Line MDA-MB 231 Is Specifically Inhibited by the Ionophore Salinomycin. Anticancer Res. 2019, 39, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.N.; Bahnassy, A.; Mourad, M.; Malash, I.; Ahmed, O.; Abdellateif, M.S. Genetic profiling of different phenotypic subsets of breast cancer stem cells (BCSCs) in breast cancer patients. Cancer Cell Int. 2022, 22, 423. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhong, X.; Qiu, Y.; Yang, L.; Wei, B.; Zhang, Z.; Bu, H. CD49f Can Act as a Biomarker for Local or Distant Recurrence in Breast Cancer. J. Breast Cancer 2017, 20, 142–149. [Google Scholar] [CrossRef]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef]
- Liu, T.; Li, K.; Zhang, Z.; Peng, J.; Yang, J.; Law, B.Y.K.; Liu, X.; Li, W. Tetrandrine Inhibits Cancer Stem Cell Characteristics and Epithelial to Mesenchymal Transition in Triple-Negative Breast Cancer via SOD1/ROS Signaling Pathway. Am. J. Chin. Med. 2023, 51, 425–444. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Grasset, E.M.; Dunworth, M.; Sharma, G.; Loth, M.; Tandurella, J.; Cimino-Mathews, A.; Gentz, M.; Bracht, S.; Haynes, M.; Fertig, E.J.; et al. Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Sci. Transl. Med. 2022, 14, eabn7571. [Google Scholar] [CrossRef]
- Yamashita, N.; Kufe, D. Addiction of Cancer Stem Cells to MUC1-C in Triple-Negative Breast Cancer Progression. Int. J. Mol. Sci. 2022, 23, 8219. [Google Scholar] [CrossRef]
- Fujisue, M.; Nishimura, R.; Okumura, Y.; Tashima, R.; Nishiyama, Y.; Osako, T.; Toyozumi, Y.; Arima, N. Clinical Significance of CK19 Negative Breast Cancer. Cancers 2012, 5, 1–11. [Google Scholar] [CrossRef]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer. 2010, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Espada, J.; Peinado, H.; Lopez-Serra, L.; Setién, F.; Lopez-Serra, P.; Portela, A.; Renart, J.; Carrasco, E.; Calvo, M.; Juarranz, A.; et al. Regulation of SNAIL1 and E-cadherin function by DNMT1 in a DNA methylation-independent context. Nucleic Acids Res. 2011, 39, 9194–9205. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Joshi, M.B. Comprehensive analysis of regulation of DNA methyltransferase isoforms in human breast tumors. J. Cancer Res. Clin. Oncol. 2021, 147, 937–971. [Google Scholar] [CrossRef]
- Alaaeldin, R.; Ali, F.E.M.; Bekhit, A.A.; Zhao, Q.L.; Fathy, M. Inhibition of NF-kB/IL-6/JAK2/STAT3 Pathway and Epithelial-Mesenchymal Transition in Breast Cancer Cells by Azilsartan. Molecules 2022, 27, 7825. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, P.G. Caveolin-1, antiapoptosis signaling, and anchorage-independent cell growth. Focus on “Caveolin-1 regulates Mcl-1 stability and anoikis in lung carcinoma cells”. Am. J. Physiol. Cell Physiol. 2012, 302, C1282–C1283. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, C.; Chen, Y.; Zhang, W.; Fu, Z.; Li, J.; Zheng, J.; Xie, M. A novel risk model based on anoikis: Predicting prognosis and immune infiltration in cutaneous melanoma. Front. Pharmacol. 2023, 13, 1090857. [Google Scholar] [CrossRef]
- Nakagawa, H.; Higurashi, M.; Ishikawa, F.; Mori, K.; Shibanuma, M. An indispensable role of TAZ in anoikis resistance promoted by OTUB1 deubiquitinating enzyme in basal-like triple-negative breast cancer cells. Biochem. Biophys. Res. Commun. 2023, 649, 1–9. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit. Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef]
- Meister, M.T.; Boedicker, C.; Linder, B.; Kögel, D.; Klingebiel, T.; Fulda, S. Concomitant targeting of Hedgehog signaling and MCL-1 synergistically induces cell death in Hedgehog-driven cancer cells. Cancer Lett. 2019, 465, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, A.; Zijlstra, A.; Zutter, M.M. Flipping the switch: Integrin switching provides metastatic competence. Sci. Signal. 2014, 7, pe9. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Li, M.; Wu, X.; Setrerrahmane, S.; Xu, H. Integrins as attractive targets for cancer therapeutics. Acta Pharm. Sin. B 2021, 11, 2726–2737. [Google Scholar] [CrossRef]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293. [Google Scholar] [CrossRef]
- Yin, H.L.; Wu, C.C.; Lin, C.H.; Chai, C.Y.; Hou, M.F.; Chang, S.J.; Tsai, H.P.; Hung, W.C.; Pan, M.R.; Luo, C.W. β1 Integrin as a Prognostic and Predictive Marker in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1432. [Google Scholar] [CrossRef] [PubMed]
- Alday-Parejo, B.; Stupp, R.; Rüegg, C. Are Integrins Still Practicable Targets for Anti-Cancer Therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef]
- Cerqueira, O.L.D.; Botelho, M.C.S.; Fiore, A.P.Z.P.; Osório, C.A.B.T.; Tomasin, R.; Morais, M.C.C.; López, R.V.M.; Cardoso, E.C.; Vilella-Arias, S.A.; Reis, E.M.; et al. Prognostic value of integrin αV expression and localization pattern in invasive breast carcinomas. Neoplasia 2022, 30, 100803. [Google Scholar] [CrossRef]
- Mian, M.F.; Kang, C.; Lee, S.; Choi, J.H.; Bae, S.S.; Kim, S.H.; Kim, Y.H.; Ryu, S.H.; Suh, P.G.; Kim, J.S.; et al. Cleavage of focal adhesion kinase is an early marker and modulator of oxidative stress-induced apoptosis. Chem. Biol. Interact. 2008, 171, 57–66. [Google Scholar] [CrossRef]
- Seo, J.; Park, M.; Ko, D.; Kim, S.; Park, J.M.; Park, S.; Nam, K.D.; Farrand, L.; Yang, J.; Seok, C.; et al. Ebastine impairs metastatic spread in triple-negative breast cancer by targeting focal adhesion kinase. Cell. Mol. Life Sci. 2023, 80, 132. [Google Scholar] [CrossRef]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef]
- Jiang, H.; Li, H. Prognostic values of tumoral MMP2 and MMP9 overexpression in breast cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 149. [Google Scholar] [CrossRef]
- Lodillinsky, C.; Fuhrmann, L.; Irondelle, M.; Pylypenko, O.; Li, X.Y.; Bonsang-Kitzis, H.; Reyal, F.; Vacher, S.; Calmel, C.; De Wever, O.; et al. Metastasis-suppressor NME1 controls the invasive switch of breast cancer by regulating MT1-MMP surface clearance. Oncogene 2021, 40, 4019–4032. [Google Scholar] [CrossRef]
- Peeney, D.; Fan, Y.; Nguyen, T.; Meerzaman, D.; Stetler-Stevenson, W.G. Matrisome-Associated Gene Expression Patterns Correlating with TIMP2 in Cancer. Sci. Rep. 2019, 9, 20142. [Google Scholar] [CrossRef]
- Ayla, S.; Karahüseyinogluc, S. Cancer Stem Cells, Their Microenvironment and Anoikis. Crit. Rev. Oncog. 2019, 24, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aliarab, A.; Asghari Vostakolaei, M.; Hotelchi, M.; Neisari, R.; Gholizadeh-Ghaleh Aziz, S.; Bazl, M.R. Crosslink between p53 and metastasis: Focus on epithelial-mesenchymal transition, cancer stem cell, angiogenesis, autophagy, and anoikis. Mol. Biol. Rep. 2021, 48, 7545–7557. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Z.Y.; Chen, L.; Zhang, J.Y.; Fu, L.Y.; Tao, L.; Zhang, Y.; Hu, X.X.; Shen, X.C. Shikonin inhibits triple-negative breast cancer-cell metastasis by reversing the epithelial-to-mesenchymal transition via glycogen synthase kinase 3β-regulated suppression of β-catenin signaling. Biochem. Pharmacol. 2019, 166, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Alsharif, S.; Sharma, P.; Bursch, K.; Milliken, R.; Lam, V.; Fallatah, A.; Phan, T.; Collins, M.; Dohlman, P.; Tiufekchiev, S.; et al. Keratin 19 maintains E-cadherin localization at the cell surface and stabilizes cell-cell adhesion of MCF7 cells. Cell Adh. Migr. 2021, 15, 1–17. [Google Scholar] [CrossRef]
- Tavakolian, S.; Goudarzi, H.; Faghihloo, E. E-cadherin, Snail, ZEB-1, DNMT1, DNMT3A and DNMT3B expression in normal and breast cancer tissues. Acta Biochim. Pol. 2019, 66, 409–414. [Google Scholar] [CrossRef]
- Fukagawa, A.; Ishii, H.; Miyazawa, K.; Saitoh, M. δEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Med. 2015, 4, 125–135. [Google Scholar] [CrossRef]
- Yang, H.L.; Thiyagarajan, V.; Shen, P.C.; Mathew, D.C.; Lin, K.Y.; Liao, J.W.; Hseu, Y.C. Anti-EMT properties of CoQ0 attributed to PI3K/AKT/NFKB/MMP-9 signaling pathway through ROS-mediated apoptosis. J. Exp. Clin. Cancer Res. 2019, 38, 186. [Google Scholar] [CrossRef] [PubMed]
- Vijay, G.V.; Zhao, N.; Den Hollander, P.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; Chang, Y.W.; Hong, M.X.; Hsu, T.C.; Lee, K.C.; Lin, C.; Lee, J.L. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT-MET switch and cancer metastasis. Oncogene 2021, 40, 791–805. [Google Scholar] [CrossRef]
- Cheng, X.; Shen, T.; Liu, P.; Fang, S.; Yang, Z.; Li, Y.; Dong, J. mir-145-5p is a suppressor of colorectal cancer at early stage, while promotes colorectal cancer metastasis at late stage through regulating AKT signaling evoked EMT-mediated anoikis. BMC Cancer 2022, 22, 1151. [Google Scholar] [CrossRef] [PubMed]
- Putri, H.E.; Sritularak, B.; Chanvorachote, P. Pongamol Inhibits Epithelial to Mesenchymal Transition Through Suppression of FAK/Akt-mTOR Signaling. Anticancer Res. 2021, 41, 6147–6154. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Park, D.; Magis, A.T.; Behera, M.; Ramalingam, S.S.; Owonikoko, T.K.; Sica, G.L.; Ye, K.; Zhang, C.; Chen, Z.; et al. Mcl-1 Interacts with Akt to Promote Lung Cancer Progression. Cancer Res. 2019, 79, 6126–6138. [Google Scholar] [CrossRef]
- Pratelli, G.; Di Liberto, D.; Carlisi, D.; Emanuele, S.; Giuliano, M.; Notaro, A.; De Blasio, A.; Calvaruso, G.; D’Anneo, A.; Lauricella, M. Hypertrophy and ER Stress Induced by Palmitate Are Counteracted by Mango Peel and Seed Extracts in 3T3-L1 Adipocytes. Int. J. Mol. Sci. 2023, 24, 5419. [Google Scholar] [CrossRef]
- De Blasio, A.; Di Fiore, R.; Morreale, M.; Carlisi, D.; Drago-Ferrante, R.; Montalbano, M.; Scerri, C.; Tesoriere, G.; Vento, R. Unusual roles of caspase-8 in triple-negative breast cancer cell line MDA-MB-231. Int. J. Oncol. 2016, 48, 2339–2348. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratelli, G.; Carlisi, D.; Di Liberto, D.; Notaro, A.; Giuliano, M.; D’Anneo, A.; Lauricella, M.; Emanuele, S.; Calvaruso, G.; De Blasio, A. MCL1 Inhibition Overcomes the Aggressiveness Features of Triple-Negative Breast Cancer MDA-MB-231 Cells. Int. J. Mol. Sci. 2023, 24, 11149. https://doi.org/10.3390/ijms241311149
Pratelli G, Carlisi D, Di Liberto D, Notaro A, Giuliano M, D’Anneo A, Lauricella M, Emanuele S, Calvaruso G, De Blasio A. MCL1 Inhibition Overcomes the Aggressiveness Features of Triple-Negative Breast Cancer MDA-MB-231 Cells. International Journal of Molecular Sciences. 2023; 24(13):11149. https://doi.org/10.3390/ijms241311149
Chicago/Turabian StylePratelli, Giovanni, Daniela Carlisi, Diana Di Liberto, Antonietta Notaro, Michela Giuliano, Antonella D’Anneo, Marianna Lauricella, Sonia Emanuele, Giuseppe Calvaruso, and Anna De Blasio. 2023. "MCL1 Inhibition Overcomes the Aggressiveness Features of Triple-Negative Breast Cancer MDA-MB-231 Cells" International Journal of Molecular Sciences 24, no. 13: 11149. https://doi.org/10.3390/ijms241311149
APA StylePratelli, G., Carlisi, D., Di Liberto, D., Notaro, A., Giuliano, M., D’Anneo, A., Lauricella, M., Emanuele, S., Calvaruso, G., & De Blasio, A. (2023). MCL1 Inhibition Overcomes the Aggressiveness Features of Triple-Negative Breast Cancer MDA-MB-231 Cells. International Journal of Molecular Sciences, 24(13), 11149. https://doi.org/10.3390/ijms241311149