
Treatment-Resistant Depression (TRD): Is the Opioid System Involved?

Abstract
1. Introduction
2. Results
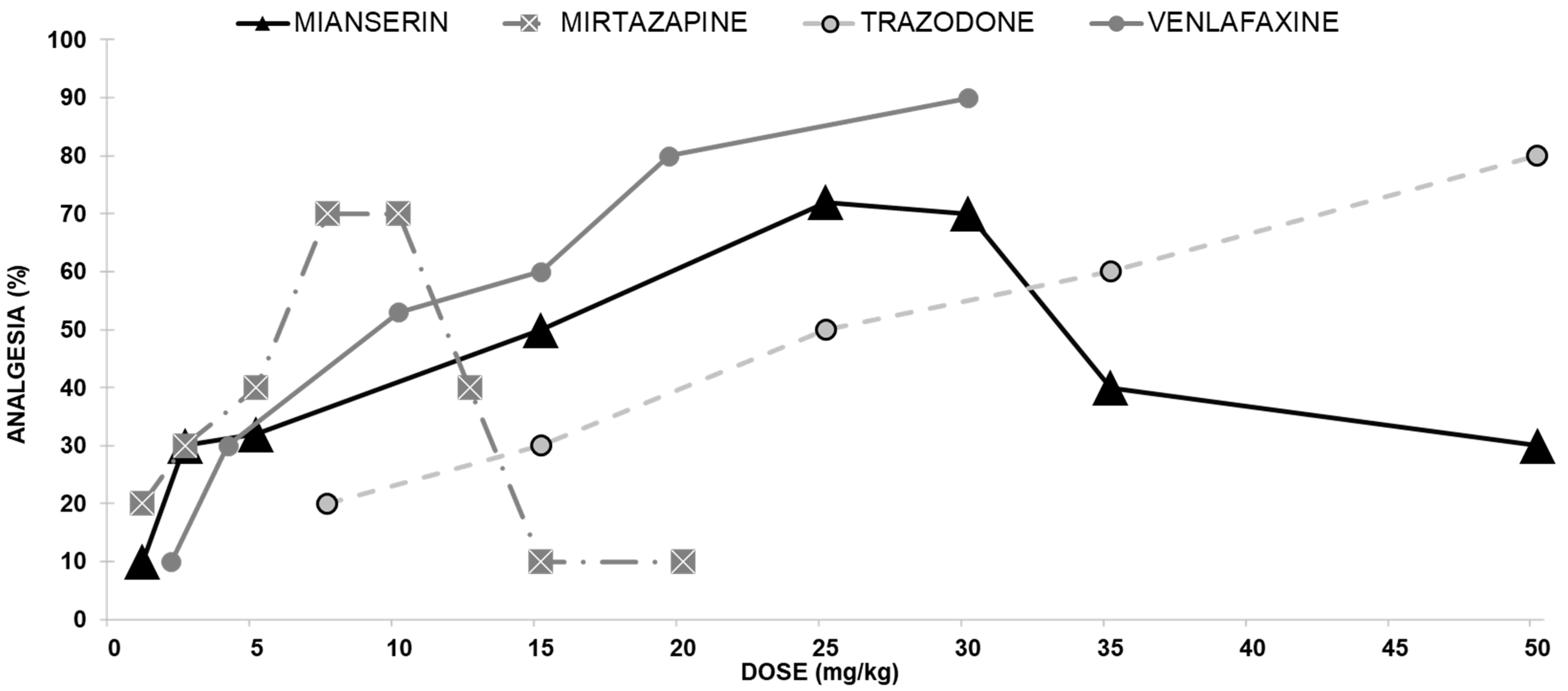
2.1. Dose-Response Curves
2.1.1. Antidepressants with Opioid Interaction
Mianserin Antinociceptive Effect
Mirtazapine Antinociceptive Effect
Trazodone Antinociceptive Effect
Venlafaxine Antinociceptive Effect
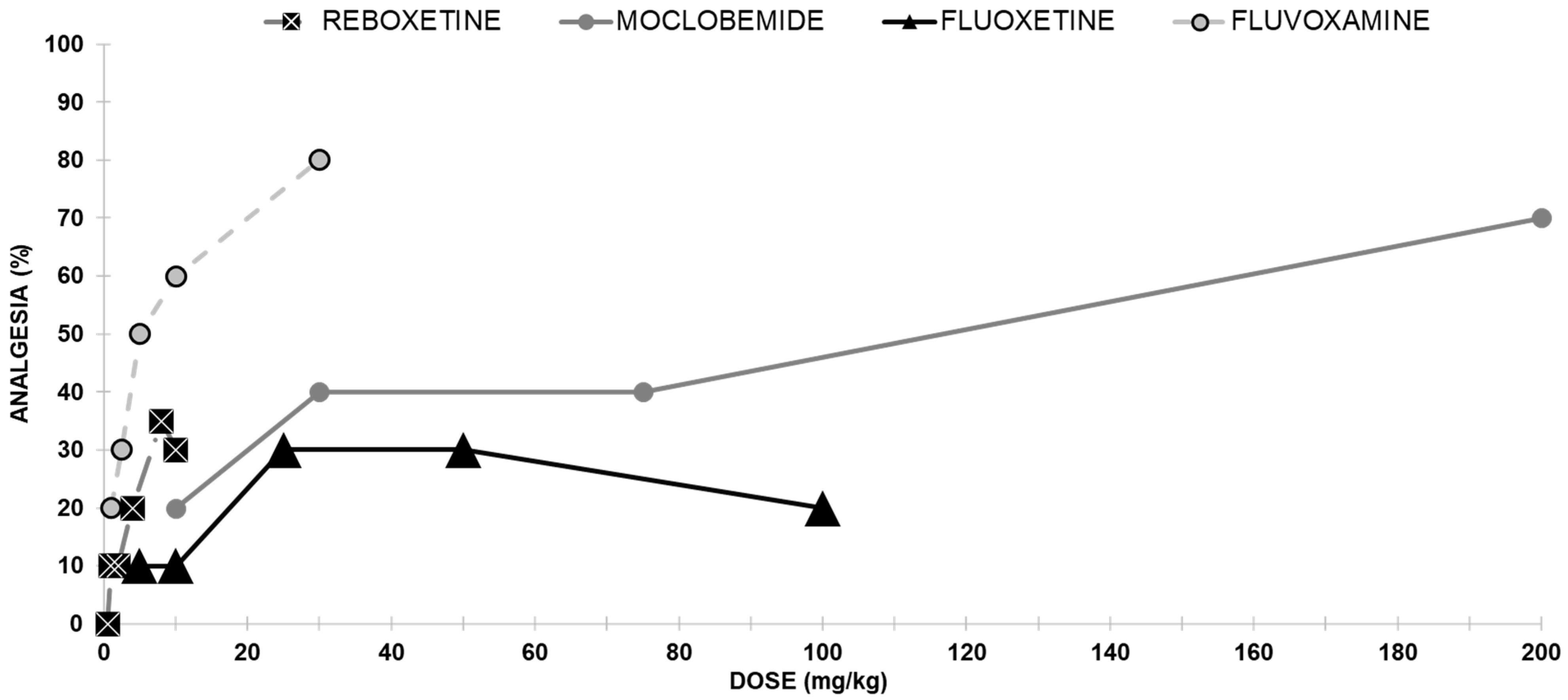
2.1.2. Antidepressants with No Opioid Interaction
Reboxetine Antinociceptive Effect
Moclobemide Antinociceptive Effect
Fluoxetine Antinociceptive Effects
Fluvoxamine Antinociceptive Effect
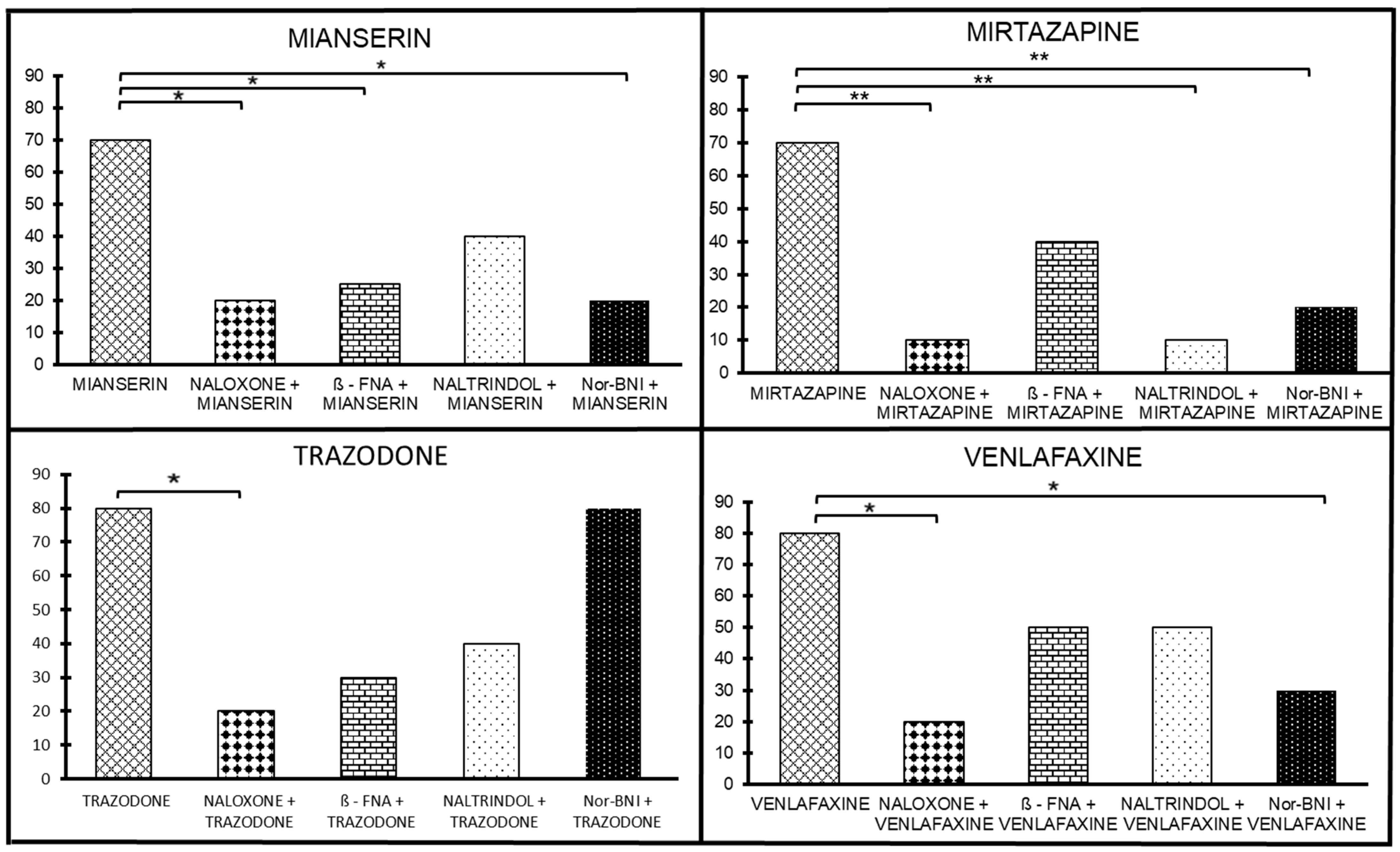
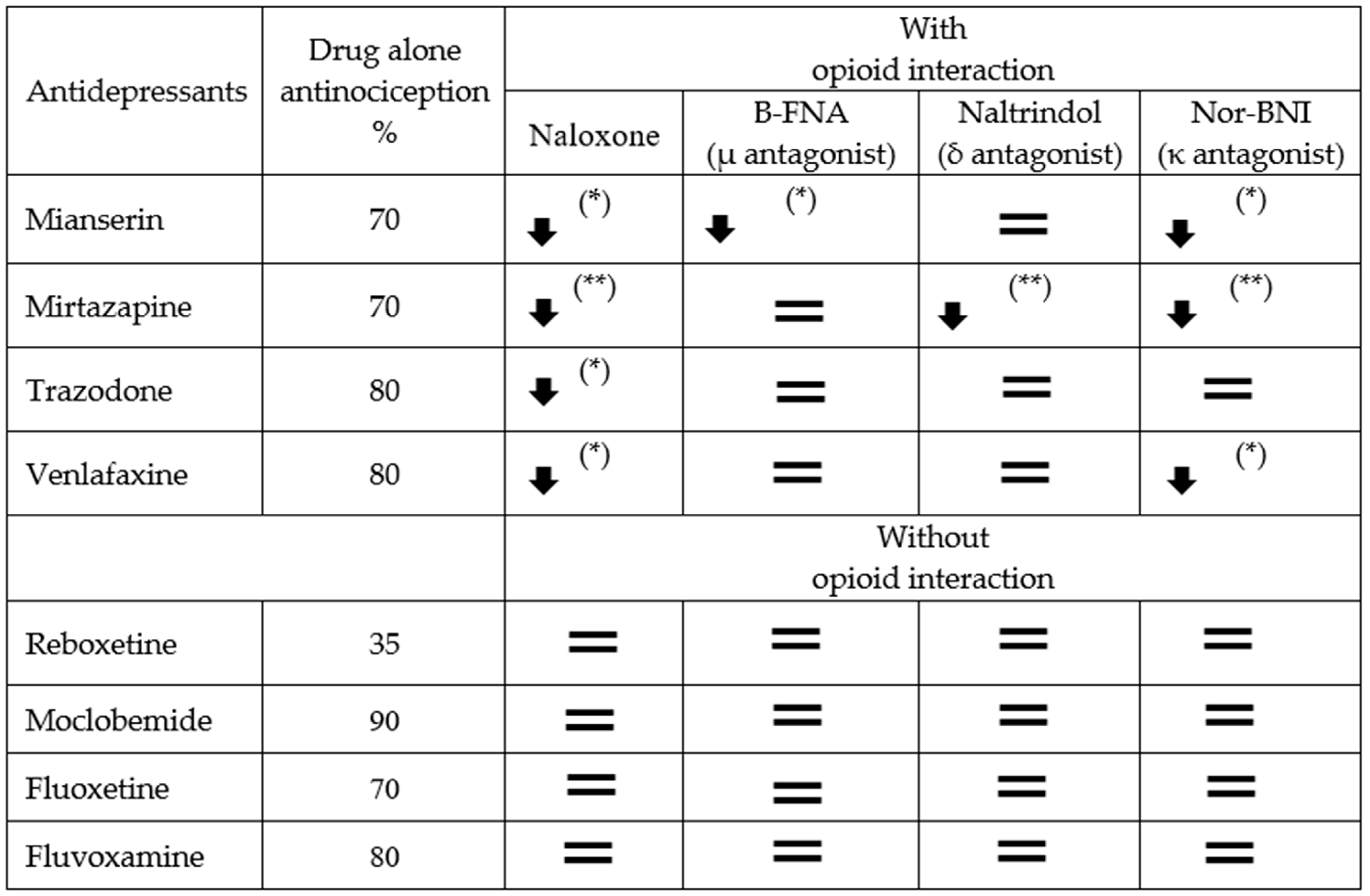
2.2. The Sensitivity of Antidepressant Drugs to the Antinociceptive Effect of Selective Opioid Receptor Antagonists
2.2.1. Antidepressants with Opioid Interaction
The Sensitivity of Mianserin Antinociceptive Effect to Selective Opioid Receptor Antagonists
The Sensitivity of the Mirtazapine Antinociceptive Effect to Selective Opioid Receptor Antagonists
The Sensitivity of the Trazodone Antinociceptive Effect to Selective Opioid Receptor Antagonists
The Sensitivity of Venlafaxine Antinociceptive Effect on Selective Opioid Receptor Antagonists
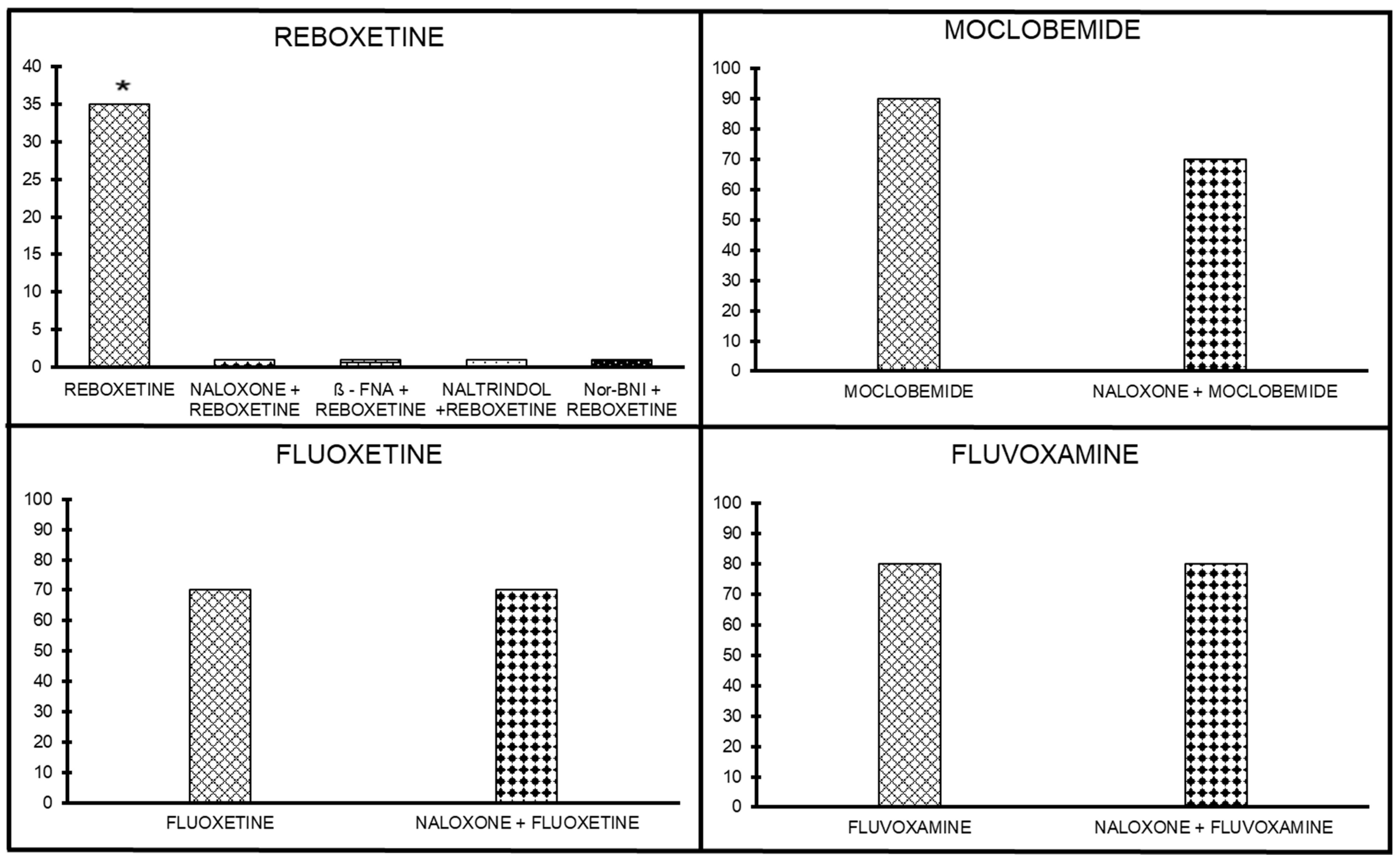
2.2.2. Antidepressants without Opioid Interaction
Antinociceptive Effect of Opioid Antagonists on Reboxetine Analgesia
Antinociceptive Effect of Opioid Antagonists on Moclobemide Analgesia
Antinociceptive Effect of Opioid Antagonists on Fluoxetine Analgesia
Antinociceptive Effect of Opioid Antagonists on Fluvoxamine Analgesia
2.3. The Sensitivity of Antidepressant Drugs to the Antinociceptive Effect of Selective Opioid Receptor Agonists
2.3.1. Antidepressants with Opioid Interaction
Mianserin Action on Selected Opioid Receptor Subtypes: Agonists
Mirtazapine Action on Selected Opioid Receptor Subtypes: Agonists
Trazodone Action on Selected Opioid Receptor Subtypes: Agonists
Venlafaxine Action on Selected Opioid Receptor Subtypes: Agonists
2.3.2. Antidepressants without Opioid Interaction
Reboxetine Action on Selected Opioid Receptor Subtypes: Agonists
Moclobemide Action on Selected Opioid Receptor Subtypes: Agonists
Fluvoxamine Action on Selected Opioid Receptor Subtypes
Fluoxetine Action on Selected Opioid Receptor Subtypes
3. Discussion
3.1. Limitations
3.2. Strengths
4. Materials and Methods
4.1. Subjects and Surgery
4.2. Agents
4.3. Antinociception Assessment
4.3.1. Hot Plate
4.3.2. Tail-Flick
4.4. Procedure
4.4.1. Experiment 1
4.4.2. Experiment 2
4.4.3. Experiment 3
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.D.; et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef]
- Hindmarch, I. Beyond the monoamine hypothesis: Mechanisms, molecules and methods. Eur. Psychiatry 2002, 17 (Suppl. S3), 249–294. [Google Scholar] [CrossRef] [PubMed]
- Little, A. Treatment-resistant depression. Am. Fam. Physician 2009, 80, 167–172. [Google Scholar]
- Baig-Ward, K.M.; Jha, M.K.; Trivedi, M.H. The Individual and Societal Burden of Treatment-Resistant Depression: An Overview. Psychiatr. Clin. N. Am. 2023, 46, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.; Cars, T.; Lööv, S.Å.; Söderling, J.; Sundström, J.; Tiihonen, J.; Leval, A.; Gannedahl, A.; Björkholm, C.; Själin, M.; et al. Association of Treatment-Resistant Depression With Patient Outcomes and Health Care Resource Utilization in a Population-Wide Study. JAMA Psychiatry 2023, 80, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.M.; Emrich, H.M. Current and historical concepts of opiate treatment in psychiatric disorders. Int. Clin. Psychopharmacol. 1988, 3, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Peter, L.; Tenore, M.D. Psychotherapeutic benefits of opioid agonist therapy. J. Addict. Dis. 2008, 27, 49–65. [Google Scholar] [CrossRef]
- Jelen, L.A.; Stone, J.M.; Young, A.H.; Mehta, M.A. The opioid system in depression. Neurosci. Biobehav. Rev. 2022, 140, 104800. [Google Scholar] [CrossRef]
- White, W.L. Slaying the Dragon: The History of Addiction Treatment and Recovery in America, 2nd ed.; Chestnut Health Syst. Maryland (USA): Maryville, IL, USA, 2014. [Google Scholar]
- Miron, O.; Zeltzer, D.; Shir, T.; Balicer, R.D.; Einav, L.; Feldman, B.S. Rising opioid prescription fulfillment among non-cancer and non-elderly patients-Israel’s alarming example. Reg. Anesth. Pain Med. 2021, 46, 455–456. [Google Scholar] [CrossRef]
- Shapira, B.; Berkovitz, R.; Haklai, Z.; Goldberger, N.; Lipshitz, I.; Rosca, P. Trends and correlated outcomes in population-level prescription opioid and transdermal fentanyl use in Israel. Isr. J. Health Policy Res. 2023, 12, 9. [Google Scholar] [CrossRef]
- Borsodi, A.; Bruchas, M.; Caló, G.; Chavkin, C.; Christie, M.J.; Civelli, O.; Connor, M.; Cox, B.M.; Devi, L.A.; Evans, C.; et al. Opioid receptors in GtoPdb v.2023.1. IUPHAR/BPS Guide Pharmacol. CITE 2023, 2023. [Google Scholar] [CrossRef]
- Berrocoso, E.; Sanchez-Blazquez, P.; Garzon, J.; Mico, A.J. Opiates as Antidepressants. Curr. Pharm. Des. 2009, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Peciña, M.; Karp, J.F.; Mathew, S.; Todtenkopf, M.S.; Ehrich, E.W.; Zubieta, J.K. Endogenous opioid system dysregulation in depression: Implications for new therapeutic approaches. Mol. Psychiatry 2019, 24, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Onali, P.; Dedoni, S.; Olianas, M.C. Direct agonist activity of tricyclic antidepressants at distinct opioid receptor subtypes. J. Pharmacol. Exp. Ther. 2010, 332, 255–265. [Google Scholar] [CrossRef]
- Besson, A.; Privat, A.M.; Eschalier, A.; Fialip, J. Dopaminergic and opioidergic mediations of tricyclic antidepressants in the learned helplessness paradigm. Pharmacol. Biochem. Behav. 1999, 64, 541–548. [Google Scholar] [CrossRef]
- Schmidt, A.W.; Peroutka, S.J. 5-hydroxytryptamine receptor “families”. FASEB J. 1989, 3, 2242–2249. [Google Scholar] [CrossRef]
- Schofield, P.R.; Shivers, B.D.; Seeburg, P. The role of receptor subtype diversity in the central nervous system. TINS 1990, 13, 8–11. [Google Scholar]
- Fineberg, N.; Montgomery, S.A. Obsessive compulsive disorder: A specific serotonergic illness? In Serotonin, Sleep and Mental Disorder; Idzikiwski, C., Cowen, P.J., Eds.; Wrightson Biomedical: London, UK, 1991; pp. 131–145. [Google Scholar]
- Pinder, R.M.; Fink, M. Mianserin. Mod. Probl. Pharmacopsychiat. 1982, 18, 70–101. [Google Scholar]
- Kopera, H. Lack of anticholinergic and cardiovascular effects of mianserin; studies in healthy subjects and heart patients. Acta Psychiatr. Scand. 1983, 67 (Suppl. S302), 81–89. [Google Scholar] [CrossRef]
- Besson, A.; Haddjeri, N.; Blier, P.; de Montigny, C. Effects of the co-administration of mirtazapine and paroxetine on serotonergic neurotransmission in the rat brain. Eur. Neuropsychopharmacol. 2000, 10, 177–188. [Google Scholar] [CrossRef]
- De Boer, T. The pharmacologic profile of mirtazapine. J. Clin. Psychiatry 2000, 57 (Suppl. S4), 19–24. [Google Scholar]
- Schreiber, S.; Backer, M.M.; Kaufman, J.; Rigai, T.; Pick, C.G. Interaction between the tetracyclic antidepressant Mianserin HCl and opioid receptors. Eur. Neuropsychopharmacol. 1998, 8, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Trazodone: A review of its pharmacological properties and therapeutic use in depression and anxiety. Drugs 1981, 21, 401–429. [Google Scholar] [CrossRef] [PubMed]
- Clements-Jewery, S.; Robson, P.A.; Chidley, L.J. Biochemical investigations into the mode of action of trazodone. Neuropharmacology 1980, 19, 1165–1173. [Google Scholar] [CrossRef]
- Clements-Jewery, S. The development of cortical b-adrenoreceptor subsensitivity in the rat by chronic treatment with trazodone, doxepine and mianserin. Neuropharmacology 1978, 17, 779–781. [Google Scholar] [CrossRef]
- Valeri, P.; Palmery, M.; Silverstrini, B. Binding profile of trazodone and dapiprazole to same brain receptors. Drugs Exp. Clin. Res. 1988, 24, 53–58. [Google Scholar]
- Lloyd, G.K.; Cronin, S.; Fletcher, A.; Mitchell, P.J. The profile of venlafaxine, a novel antidepressant agent, in behavioral antidepressant drug models. Clin. Neuropharm. 1992, 15 Pt B, 428B. [Google Scholar] [CrossRef]
- Moyer, J.A.; Andree, T.H.; Haskins, J.T.; Husbands, G.E.M.; Muth, E.A. The preclinical pharmacological profile of venlafaxine: A novel antidepressant agent. Clin. Neuropharm. 1992, 15 Pt B, 435B. [Google Scholar] [CrossRef]
- Muth, E.A.; Haskins, J.T.; Moyer, J.A.; Husbands, G.E.M.; Nielsen, S.T.; Sigg, E.B. Antidepressant biochemical profile of the novel bicyclic compound Wy-45,030, an ethyl cyclohezanol derivative. Biochem. Pharmacol. 1986, 35, 4493–4497. [Google Scholar] [CrossRef]
- Mendlewicz, J. Pharmacologic profile and efficacy of venlafaxine. Int. Clin. Psychopharmacol. 1995, 10 (Suppl. S2), 5–13. [Google Scholar] [CrossRef]
- Ellingrod, V.L.; Perry, P.J. Venlafaxine: A heterocyclic antidepressant. Am. J. Hosp. Pharm. 1994, 51, 3030–3046. [Google Scholar] [CrossRef]
- Richelson, E. Synaptic effects of antidepressants. J. Clin. Psychopharmacol. 1986, 16 (Suppl. S2), 1S–9S. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.J.; Spencer, C.M. Reboxetine. CNS Drugs 1999, 12, 65–83. [Google Scholar] [CrossRef]
- Bernstein, J.G. Monoamine oxidase inhibitors. In Handbook of Drug Therapy in Psychiatry, 2nd ed.; Bernstein, J.G., Ed.; PSG Publishing Company: Littleton, MA, USA, 1988; pp. 161–188. [Google Scholar]
- Paykel, E.S. Clinical efficacy of reversible and selective inhibitors of monoamine oxidase A in major depression. Acta Psychiatr. Scand. 1995, 91, 22–27. [Google Scholar]
- Priest, R.G. Moclobemide: A range of opportunities. Psychopharmacology 1992, 106, 5140–5142. [Google Scholar] [CrossRef]
- Hofmann, F. (Ed.) Product Information; La Roche: Basel, Switzerland, 1994. [Google Scholar]
- Leonard, B.E. Pharmacological differences of serotonin reuptake inhibitors and possible clinical relevance. Drugs 1992, 43 (Suppl. S2), 3–10. [Google Scholar] [CrossRef]
- Stahl, S.M. Not so selective serotonin reuptake inhibitors. J. Clin. Psychiatry 1998, 59, 343–344. [Google Scholar] [CrossRef]
- Chen, F.; Larsen, M.B.; Sanchez, C.; Wiborg, O. The Senantiomer of R,S-citalopram, increases inhibitor binding to the human serotonin transporter by an allosteric mechanism. Comparison with other serotonin transporter inhibitors. Eur. Neuropsychopharmacol. 2005, 15, 193–198. [Google Scholar] [CrossRef]
- Schreiber, S.; Pick, C.G. From selective to highly-selective SSRIs: A comparison of the antinociceptive properties of fluoxetine, fluvoxamine, citalopram and escitalopram. Eur. Neuropsychopharmacol. 2006, 16, 464–468. [Google Scholar] [CrossRef]
- Ban, T.A. Pharmacotherapy of depression: A historical analysis. J. Neural. Trans. 2001, 108, 707–716. [Google Scholar] [CrossRef]
- Nyro, G. Pszichiatria. Medicina 1962, Budapest. Available online: https://moly.hu/konyvek/nyiro-gyula-szerk-psychiatria (accessed on 20 May 2023).
- Davis, J.M.; Wang, Z.; Janicak, P.G. A quantitative analysis of clinical drug trials for the treatment of affective disorders. Psychopharmacol. Bull. 1993, 129, 175–181. [Google Scholar]
- Ban, T.A. selection of pharmacological treatment of depressive illness, part two. Efficacy and differential activity of antidepressants. Neuropsychopharmacol. Hung. 1999, 2, 3–8. [Google Scholar]
- Fritze, J. The adrenergic-cholinergic imbalance hypothesis of depression: A review and a perspective. Rev. Neurosci. 1993, 4, 63–93. [Google Scholar] [CrossRef]
- O’Brien, C.P.; Volpicelli, L.A.; Volpicelli, J.R. Naltrexone in the treatment of alcoholism: A clinical review. Alcohol 1996, 13, 35–39. [Google Scholar] [CrossRef]
- Amiaz, R.; Stein, O.; Dannon, P.N.; Grunhaus, L.; Schreiber, S. Resolution of treatment-refractory depression with naltrexone augmentation of paroxetine—A case report. Psychopharmacology 1999, 143, 433–434. [Google Scholar] [CrossRef]
- O’Mara, N.B.; Wesley, L.C. Naltrexone in the treatment of alcohol dependence. Ann. Pharmacother. 1994, 28, 210–211. [Google Scholar]
- Shufman, E.N.; Porat, S.; Witztum, E.; Gandaeu, D.; Bar-Hamburger, R.; Ginath, Y. The efficacy of naltrexone in preventing re-abuse of heroin after detoxification. Biol. Psychiatry 1994, 35, 935–945. [Google Scholar] [CrossRef]
- Mitchell, J.E.; Raymond, N.; Specker, S. A review of the controlled trials of pharmacotherapy and psychotherapy in the treatment of bulimia nervosa. Int. J. Eat. Disord. 1993, 143, 229–247. [Google Scholar] [CrossRef]
- Benjamin, E.; Bout-Smith, T. Naltrexone and fuoxetine in Prader-Willi syndrome. J. Am. Acad. Child Adolesc. Psychiatry 1993, 32, 870–873. [Google Scholar] [CrossRef]
- Ahee, L.; Attila, L.M.; Carlson, K.R. Augmentation of morphine induced changes in brain monoamine metabolism after chronic naltrexone treatment. J. Pharmacol. Exp. Ther. 1990, 255, 803–808. [Google Scholar]
- Riblet, N.B.; Young-Xu, Y.; Shiner, B.; Schnurr, P.P.; Watts, B.V. The efficacy and safety of buprenorphine for the treatment of depression: A systematic review and meta-analysis. J. Psychiatr. Res. 2023, 161, 393–401. [Google Scholar] [CrossRef]
- Pick, C.G.; Peter, Y.; Schreiber, S.; Weizman, R. Pharmacological characterization of buprenorphine, a mixed agonist-antagonist with kappa3 analgesia. Brain Res. 1997, 744, 41–46. [Google Scholar] [CrossRef]
- Smith, K.M.; Nguyen, E.; Ross, S.E. The Delta-Opioid Receptor Bidirectionally Modulates Itch. J. Pain 2023, 24, 264–272. [Google Scholar] [CrossRef]
- Lutfy, K.; Cowan, A. Buprenorphine: A unique drug with complex pharmacology. Curr. Neuropharmacol. 2004, 2, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Bastien, G.; McAnulty, C.; Ledjiar, O.; Socias, M.E.; Le Foll, B.; Lim, R.; Hassan, A.N.; Brissette, S.; Marsan, S.; Talbot, A.; et al. OPTIMA Research Group within the Canadian Research Initiative in Substance Misuse. Effects of Buprenorphine/Naloxone and Methadone on Depressive Symptoms in People with Prescription Opioid Use Disorder: A Pragmatic Randomised Controlled Trial. Can. J. Psychiatry 2022, 14, 7067437221145013. [Google Scholar] [CrossRef]
- Domani, Y.; Sharon, H.; Tarrasch, R.; Meidan, R.; Schreiber, S.; Bloch, M.; Hendler, T.; Bleich-Cohen, M. Repeated oral ketamine for ambulatory treatment of resistant depression. A randomized, double-blind, placebo-controlled proof-of-concept study. Br. J. Psychiatry 2019, 214, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.; Chen, L.N.; Lane, R.; Doherty, T.; Drevets, W.C.; Morrison, R.L.; Sanacora, G.; Wilkinson, S.T.; Popova, V.; Fu, D.J. Long-term safety and maintenance of response with esketamine nasal spray in participants with treatment-resistant depression: Interim results of the SUSTAIN-3 study. Neuropsychopharmacology 2023, 48, 1225–1233. [Google Scholar] [CrossRef]
- Zhang, F.; Hillhouse, T.M.; Anderson, P.M.; Koppenhaver, P.O.; Kegen, T.N.; Manicka, S.G.; Lane, J.T.; Pottanat, E.; Van Fossen, M.; Rice, R.; et al. Opioid receptor system contributes to the acute and sustained antidepressant-like effects, but not the hyperactivity motor effects of ketamine in mice. Pharmacol. Biochem. Behav. 2021, 208, 173228. [Google Scholar] [CrossRef]
- Faouzi, A.; Varga, B.R.; Majumdar, S. Biased Opioid Ligands. Molecules 2020, 25, 4257. [Google Scholar] [CrossRef]
- Hylden, J.L.; Wilcox, G.L. Intrathecal morphine in mice: A new technique. Eur. J. Pharmacol. 1980, 67, 313–316. [Google Scholar] [CrossRef]
- Schreiber, S.; Bleich, A.; Pick, C.G. Venlafaxine and mirtazapine: Different mechanisms of antidepressant action, common opioid-mediated antinociceptive effects—A possible opioid involvement in severe depression? J. Mol. Neurosci. 2002, 18, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Pick, C.G. Trazodone and Mirtazapine: A possible opioid involvement in their use (at low dose) for sleep? Med. Hypotheses 2020, 136, 109501. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Frishtick, R.; Volis, I.; Rubovitch, V.; Pick, C.G.; Weizman, R. The antinociceptive properties of reboxetine in acute pain. Eur. Neuropsychopharmacol. 2009, 19, 735–739. [Google Scholar] [CrossRef]
- Schreiber, S.; Backer, M.M.; Weizman, R.; Pick, C.G. The antinociceptive effects of fluoxetine. Pain Clin. 1996, 9, 349–356. Available online: http://www.vsppub.com/jconts/TPC/jc-Vol9No3pp241362199.html (accessed on 20 May 2023).
- Schreiber, S.; Backer, M.M.; Yanai, J.; Pick, C.G. The antinociceptive effect of fluvoxamine. Eur. Neuropsychopharmacol. 1996, 6, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Getslev, V.; Weizman, A.; Pick, C.G. The antinociceptive effect of moclobemide in mice is mediated by noradrenergic pathways. Neurosci. Lett. 1998, 253, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Backer, M.M.; Pick, C.G. The antinociceptive effect of venlafaxine in mice is mediated through opioid and adrenergic mechanisms. Neurosci. Lett. 1999, 273, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Backer, M.M.; Herman, I.; Shamir, D.; Boniel, T.; Pick, C.G. The antinociceptive effect of trazodone in mice is mediated through both m-opioid and serotonergic mechanisms. Behav. Brain Res. 2000, 114, 51–56. [Google Scholar]
- Schreiber, S.; Rigai, T.; Katz, Y.; Pick, C.G. The antinociceptive effect of mirtazapine in mice is mediated through serotonergic, noradrenergic and opioid mechanisms. Brain Res. Bul. 2002, 58, 599–603. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIANSERIN | MIRTAZAPINE | ||||
|---|---|---|---|---|---|
| Opioid receptor subtypes: | Without MIANSERIN | With MIANSERIN | Opioid receptor subtypes: | Without MIRTAZAPINE | With MIRTAZAPINE |
| Morphine (µ-subtype) | 4.5 mg/kg (1.7, 20.7) | 0.6 mg/kg (0.04, 0.9) | Morphine (µ-subtype) | 5.3 mg/kg (3.5, 10.3) | 1.9 mg/kg (1.1, 4.5) |
| DPDPE (δ-subtype) | 298 ng (183, 540) | 132 ng (64, 294) | DPDPE (δ-subtype) | 307 ng (190.7, 537.8) | 236 ng (144.5, 440.8) |
| U50,488H (K1-subtype) | 4.8 mg/kg (2.8, 10.3) | 0.5 mg/kg (0.2, 1.2) | U50,488H (K1-subtype) | 4.4 mg/kg (2.1, 10.0) | 3.1 mg/kg (1.3, 9.8) |
| TRAZODONE | VENLAFAXINE | ||||
| Opioid receptor subtypes: | Without TRAZODONE | With TRAZODONE | Opioid receptor subtypes: | Without VENLAFAXINE | With VENLAFAXINE |
| Morphine (µ-subtype) | 5.8 mg/kg (3.5, 14.6) | 0.4 mg/kg (0.22, 0.9) | Morphine (µ-subtype) | 5.8 mg/kg (3.5, 14.2) | 1.8 mg/kg (0.9, 3.6) |
| DPDPE (δ-subtype) | 318.24 ng (181, 624) | 167.5 ng (94.8, 321.9) | DPDPE (δ-subtype) | 320 ng (178, 653) | 90 ng (49,174) |
| U50,488H (K1-subtype) | 4.4 mg/kg (2.1, 9.8) | 1.35 mg/kg (0.6, 3.2) | U50,488H (K1-subtype) | 5.7 mg/kg (2.2, 103.2) | 1.0 mg/kg (0.4, 0.4) |
| FLUOXETINE | FLUVOXAMINE | |||
|---|---|---|---|---|
| Opioid Receptor Subtypes: | Without FLUVOXAMINE | With FLUVOXAMINE | Without FLUVOXAMINE | With FLUVOXAMINE |
| Morphine (µ-subtype) | 4.9 mg/kg (2.8, 13.5) | 1.1 mg/kg (0.2, 5.0) | 3.5 mg/kg (1.8, 6.8) | 1.4 mg/kg (0.8, 2.8) |
| DPDPE (δ-subtype) | 318 ng (200, 568) | 86 ng (44, 155) | 308 ng (238, 649) | 341 ng (200, 567) |
| U50,488H (K1-subtype) | 5.1 mg/kg (2.8, 12.7) | 0.5 mg/kg (0.2, 1.2) | 4.9 mg/kg (2.8, 10.4) | 1.8 mg/kg (0.8, 4.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreiber, S.; Keidan, L.; Pick, C.G. Treatment-Resistant Depression (TRD): Is the Opioid System Involved? Int. J. Mol. Sci. 2023, 24, 11142. https://doi.org/10.3390/ijms241311142
Schreiber S, Keidan L, Pick CG. Treatment-Resistant Depression (TRD): Is the Opioid System Involved? International Journal of Molecular Sciences. 2023; 24(13):11142. https://doi.org/10.3390/ijms241311142
Chicago/Turabian StyleSchreiber, Shaul, Lee Keidan, and Chaim G. Pick. 2023. "Treatment-Resistant Depression (TRD): Is the Opioid System Involved?" International Journal of Molecular Sciences 24, no. 13: 11142. https://doi.org/10.3390/ijms241311142
APA StyleSchreiber, S., Keidan, L., & Pick, C. G. (2023). Treatment-Resistant Depression (TRD): Is the Opioid System Involved? International Journal of Molecular Sciences, 24(13), 11142. https://doi.org/10.3390/ijms241311142