

Identification and Evolutionary Analysis of Cotton (Gossypium hirsutum) WOX Family Genes and Their Potential Function in Somatic Embryogenesis

Abstract
1. Introduction
2. Results
2.1. Identification of WOX Genes in Cotton
2.2. Phylogeny Analysis of Cotton WOX Genes
2.3. Gene Structure and Conserved Amino Acid Motif Analysis of Cotton WOX Genes
2.4. Evolutionary Analysis of WOX Gene Family in Core Seed Plants
2.5. Collinearity and Duplication Analysis of Cotton WOX Genes
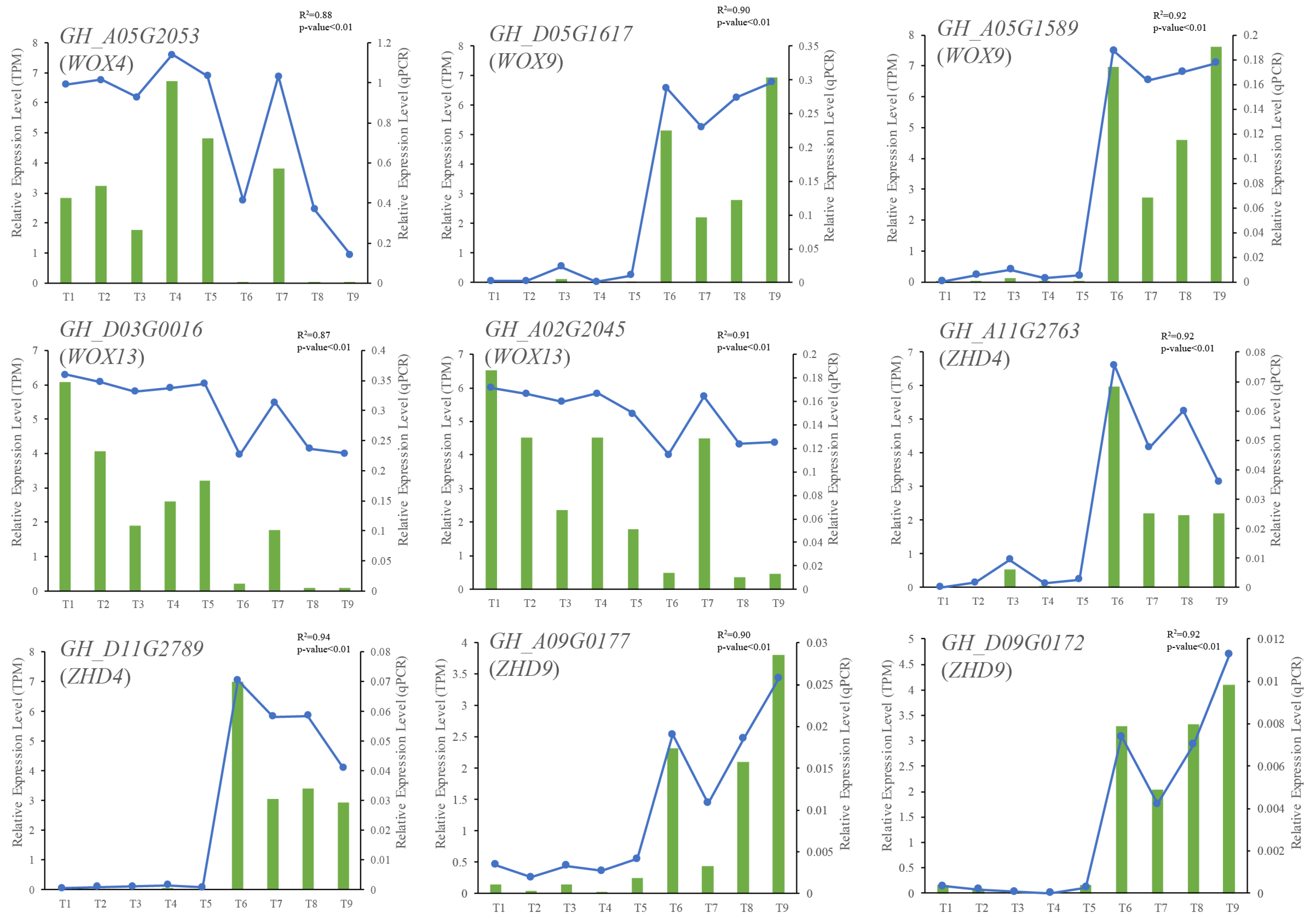
2.6. qRT-PCR Validation of RNA Sequencing
2.7. Expression Pattern of Cotton WOX Genes in Somatic Embryogenesis
2.8. Cis-Acting Regulatory Elements Analysis of Cotton WOX Genes’ Promoters
2.9. Gene Co-Expression Analysis of Cotton WOX Genes during Somatic Embryogenesis
3. Discussion
4. Materials and Methods
4.1. Identification of WOX Genes in Upland Cotton
4.2. Phylogenetic Analysis, Gene Structure, and Motif Analysis of Cotton WOX Genes
4.3. Genomic Distribution, Collinearity, and Duplication Analysis of Cotton WOX Genes
4.4. Expression Pattern Analysis of Cotton WOX Genes during Somatic Embryogenesis
4.5. qRT-PCR Validation of the RNA Sequencing
4.6. Co-Expression Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leelavathi, S.; Sunnichan, V.G.; Kumria, R.; Vijaykanth, G.P.; Bhatnagar, R.K.; Reddy, V.S. A simple and rapid Agrobacterium-mediated transformation protocol for cotton (Gossypium hirsutum L.): Embryogenic calli as a source to generate large numbers of transgenic plants. Plant Cell Rep. 2004, 22, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, M.; Li, Y.; Zhang, Q.; Lindsey, K.; Daniell, H.; Jin, S.; Zhang, X. Multi-omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation process. Plant Biotechnol. J. 2019, 17, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Deo, P.C.; Tyagi, A.P.; Taylor, M.; Harding, R.; Becker, D. Factors affecting somatic embryogenesis and transformation in modern plant breeding. S. Pac. J. Nat. App. Sci. 2011, 28, 27–40. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.; Engstrom, E.M.; Nimchuk, Z.L.; Pruneda-Paz, J.L.; Tarr, P.T.; Yan, A.; Kay, S.A.; Meyerowitz, E.M. Control of plant stem cell function by conserved interacting transcriptional regulators. Nature 2015, 517, 377–380. [Google Scholar] [CrossRef]
- Li, X.; Hamyat, M.; Liu, C.; Ahmad, S.; Gao, X.; Guo, C.; Wang, Y.; Guo, Y. Identification and Characterization of the WOX Family Genes in Five Solanaceae Species Reveal Their Conserved Roles in Peptide Signaling. Genes 2018, 9, 260. [Google Scholar] [CrossRef]
- van der Graaff, E.; Laux, T.; Rensing, S.A. The WUS homeobox-containing (WOX) protein family. Genome Biol. 2009, 10, 248. [Google Scholar] [CrossRef]
- Segatto, A.L.; Thompson, C.E.; Freitas, L.B. Molecular evolution analysis of WUSCHEL-related homeobox transcription factor family reveals functional divergence among clades in the homeobox region. Dev. Genes Evol. 2016, 226, 259–268. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, F.W.; Kramer, E.M. Large-scale phylogenomic analysis suggests three ancient superclades of the WUSCHEL-RELATED HOMEOBOX transcription factor family in plants. PLoS ONE 2019, 14, e0223521. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 2009, 21, 3493–3505. [Google Scholar] [CrossRef]
- Gallois, J.L.; Nora, F.R.; Mizukami, Y.; Sablowski, R. WUSCHEL induces shoot stem cell activity and developmental plasticity in the root meristem. Genes Dev. 2004, 18, 375–380. [Google Scholar] [CrossRef]
- Mayer, K.F.; Schoof, H.; Haecker, A.; Lenhard, M.; Jürgens, G.; Laux, T.J.C. Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 1998, 95, 805. [Google Scholar] [CrossRef] [PubMed]
- Laux; Mayer, T.; Berger, K.F.; Jurgens, J.J.D. The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development 1996, 122, 87. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Niu, Q.W.; Frugis, G.; Chua, N.H. The WUSCHEL gene promotes vegetative-to-embryonic transition in Arabidopsis. Plant J. 2002, 30, 349–359. [Google Scholar] [CrossRef]
- Wolabu, T.W.; Wang, H.; Tadesse, D.; Zhang, F.; Behzadirad, M.; Tvorogova, V.E.; Abdelmageed, H.; Liu, Y.; Chen, N.; Chen, J.; et al. WOX9 functions antagonistic to STF and LAM1 to regulate leaf blade expansion in Medicago truncatula and Nicotiana sylvestris. New Phytol. 2021, 229, 1582–1597. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Niu, L.; McHale, N.A.; Ohme-Takagi, M.; Mysore, K.S.; Tadege, M. Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc. Natl. Acad. Sci. USA 2013, 110, 366–371. [Google Scholar] [CrossRef]
- Wang, K.; Shi, L.; Liang, X.; Zhao, P.; Wang, W.; Liu, J.; Chang, Y.; Hiei, Y.; Yanagihara, C.; Du, L.; et al. The gene TaWOX5 overcomes genotype dependency in wheat genetic transformation. Nat. Plants 2022, 8, 110–117. [Google Scholar] [CrossRef]
- Hoerster, G.; Wang, N.; Ryan, L.; Wu, E.; Anand, A.; McBride, K.; Lowe, K.; Jones, T.; Gordon-Kamm, B. Use of non-integrating Zm-Wus2 vectors to enhance maize transformation. In Vitr. Cell. Dev. Biol.-Plant 2020, 56, 265–279. [Google Scholar] [CrossRef]
- Jones, T.; Lowe, K.; Hoerster, G.; Anand, A.; Gordon-Kamm, W. Maize Transformation Using the Morphogenic Genes Baby Boom and Wuschel2: Methods and Protocols. Transgenic Plants 2019, 1864, 81–93. [Google Scholar]
- Palovaara, J.; Hakman, I. Conifer WOX-related homeodomain transcription factors, developmental consideration and expression dynamic of WOX2 during Picea abies somatic embryogenesis. Plant Mol. Biol. 2008, 66, 533–549. [Google Scholar] [CrossRef]
- Palovaara, J.; Hakman, I.J.P.S. Behavior, WOX2 and polar auxin transport during spruce embryo pattern formation. Plant Signal. Behav. 2009, 4, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Singh, S.; Kaur, K.; Tiwari, S. Genome-wide identification and expression profiling of WUSCHEL-related homeobox (WOX) genes confer their roles in somatic embryogenesis, growth and abiotic stresses in banana. 3 Biotech 2022, 12, 321. [Google Scholar] [CrossRef] [PubMed]
- Bouchabke-Coussa, O.; Obellianne, M.; Linderme, D.; Montes, E.; Maia-Grondard, A.; Vilaine, F.; Pannetier, C. Wuschel overexpression promotes somatic embryogenesis and induces organogenesis in cotton (Gossypium hirsutum L.) tissues cultured in vitro. Plant Cell Rep. 2013, 32, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, X.; Yang, Z.; Wu, J.; Li, F.; Duan, L.; Liu, C.; Lu, L.; Zhang, C.; Li, F. AtWuschel promotes formation of the embryogenic callus in Gossypium hirsutum. PLoS ONE 2014, 9, e87502. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, Y.; Ding, Y.; Wu, J.; Wang, P.; Yu, Y.; Wei, X.; Wang, Y.; Zhang, C.; Li, F.; et al. Effects of GhWUS from upland cotton (Gossypium hirsutum L.) on somatic embryogenesis and shoot regeneration. Plant Sci. 2018, 270, 157–165. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, Q.; Xia, H.; Han, B.; Li, M.; Wang, Y.; Zhu, M.; Jiao, C.; Wang, D.; Zhu, J.; et al. Genome-Wide Identification and Comparative Analysis of WOX Genes in Four Euphorbiaceae Species and Their Expression Patterns in Jatropha curcas. Front. Genet. 2022, 13, 878554. [Google Scholar] [CrossRef]
- Dai, R.; Jin, H.-P.; Wang, Z.; Avihai, P.; Xu, H.-Y.; Zhang, W.; Chen, S.-W.; Ma, H.-Q. Cloning and Characterization of WOX4 Gene from Vitis vinifera L. Involved in Stem Cell Regulation. Agric. Sci. China 2011, 10, 1861–1871. [Google Scholar] [CrossRef]
- Wang, M.M.; Liu, M.M.; Ran, F.; Guo, P.C.; Ke, Y.Z.; Wu, Y.W.; Wen, J.; Li, P.F.; Li, J.N.; Du, H. Global Analysis of WOX Transcription Factor Gene Family in Brassica napus Reveals Their Stress- and Hormone-Responsive Patterns. Int. J. Mol. Sci. 2018, 19, 3470. [Google Scholar] [CrossRef]
- Yang, Z.; Gong, Q.; Qin, W.; Yang, Z.; Cheng, Y.; Lu, L.; Ge, X.; Zhang, C.; Wu, Z.; Li, F. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef]
- Breuninger, H.; Rikirsch, E.; Hermann, M.; Ueda, M.; Laux, T. Differential expression of WOX genes mediates apical-basal axis formation in the Arabidopsis embryo. Dev. Cell 2008, 14, 867–876. [Google Scholar] [CrossRef]
- Haecker, A.; Gross-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657–668. [Google Scholar] [CrossRef]
- Romera-Branchat, M.; Ripoll, J.J.; Yanofsky, M.F.; Pelaz, S. The WOX13 homeobox gene promotes replum formation in the Arabidopsis thaliana fruit. Plant J. 2013, 73, 37–49. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhang, Y.; Liu, H.; Yuan, Y.; Wang, C.; Yu, J.; Xiao, G. Comprehensive analysis of WOX genes uncovers that WOX13 is involved in phytohormone-mediated fiber development in cotton. BMC Plant Biol. 2019, 19, 312. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Shimizu, R.; Sinha, N.; Scanlon, M.J. Analyses of WOX4 transgenics provide further evidence for the evolution of the WOX gene family during the regulation of diverse stem cell functions. Plant Signal Behav. 2010, 5, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Etchells, J.P.; Provost, C.M.; Mishra, L.; Turner, S.R. WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organisation. Development 2013, 140, 2224–2234. [Google Scholar] [CrossRef]
- Lie, C.; Kelsom, C.; Wu, X. WOX2 and STIMPY-LIKE/WOX8 promote cotyledon boundary formation in Arabidopsis. Plant J. 2012, 72, 674–682. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- van Eck, N.J.; Waltman, L. Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics 2010, 84, 523–538. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Name | Domains | Clade | Gene ID | Gene Name | Domains | Clade |
|---|---|---|---|---|---|---|---|
| GH_A01G1199 | WOX4 | WUS-box/HD | Modern | GH_D01G1267 | WOX4 | WUS-box/HD | Modern |
| GH_A01G1552 | WOX1 | WUS-box/HD | Modern | GH_D01G1725 | WOX1 | WUS-box/HD | Modern |
| GH_A02G0907 | WOX4 | WUS-box/HD | Modern | GH_D02G0922 | WOX4 | WUS-box/HD | Modern |
| GH_A02G2045 | WOX13 | HD | Ancient | GH_D03G0016 | WOX13 | HD | Ancient |
| GH_A03G0470 | WOX3 | WUS-box/HD | Modern | GH_D03G1507 | WOX3 | WUS-box/HD | Modern |
| GH_A05G1005 | WOX3 | WUS-box/HD | Modern | GH_D05G0992 | WOX3 | WUS-box/HD | Modern |
| GH_A05G1589 | WOX9 | HD | Intermediate | GH_D05G1617 | WOX9 | HD | Intermediate |
| GH_A05G2053 | WOX4 | WUS-box/HD | Modern | GH_D05G2084 | WOX4 | WUS-box/HD | Modern |
| GH_A07G1065 | WOX2 | WUS-box/HD | Modern | GH_D07G1052 | WOX2 | WUS-box/HD | Modern |
| GH_A07G1886 | WOX13 | HD | Ancient | GH_D07G1885 | WOX13 | HD | Ancient |
| GH_A08G0330 | WOX8 | HD | Ancient | GH_D08G0351 | WOX8 | HD | Ancient |
| GH_A10G0308 | WOX9 | HD | Intermediate | GH_D10G0320 | WOX9 | HD | Intermediate |
| GH_A10G0983 | WUS | WUS-box/HD/EAR | Modern | GH_D10G0973 | WUS | WUS-box/HD | Modern |
| GH_A10G2581 | WOX5 | WUS-box/HD/EAR | Modern | GH_D10G2684 | WOX5 | WUS-box/HD | Modern |
| GH_A11G3426 | WOX11 | HD | Intermediate | GH_D11G3431 | WOX11 | HD | Intermediate |
| GH_A11G3685 GH_A12G0600 | WOX8 WUS | HD WUS-box/HD/EAR | Ancient Modern | GH_D11G3714 GH_D12G0619 | WOX8 WUS | HD WUS-box/HD/EAR | Ancient Modern |
| GH_A12G2894 | WOX1 | WUS-box/HD | Modern | GH_D12G2917 | WOX1 | WUS-box/HD | Modern |
| GH_A13G1885 | WOX11 | HD | Intermediate | GH_D13G1838 | WOX11 | HD | Intermediate |
| GH_A13G2563 | WOX2 | WUS-box/HD | Modern | GH_D13G2554 | WOX2 | WUS-box/HD | Modern |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Zhang, X.; Ma, D.; Liu, C. Identification and Evolutionary Analysis of Cotton (Gossypium hirsutum) WOX Family Genes and Their Potential Function in Somatic Embryogenesis. Int. J. Mol. Sci. 2023, 24, 11077. https://doi.org/10.3390/ijms241311077
Sun R, Zhang X, Ma D, Liu C. Identification and Evolutionary Analysis of Cotton (Gossypium hirsutum) WOX Family Genes and Their Potential Function in Somatic Embryogenesis. International Journal of Molecular Sciences. 2023; 24(13):11077. https://doi.org/10.3390/ijms241311077
Chicago/Turabian StyleSun, Ruibin, Xue Zhang, Dan Ma, and Chuanliang Liu. 2023. "Identification and Evolutionary Analysis of Cotton (Gossypium hirsutum) WOX Family Genes and Their Potential Function in Somatic Embryogenesis" International Journal of Molecular Sciences 24, no. 13: 11077. https://doi.org/10.3390/ijms241311077
APA StyleSun, R., Zhang, X., Ma, D., & Liu, C. (2023). Identification and Evolutionary Analysis of Cotton (Gossypium hirsutum) WOX Family Genes and Their Potential Function in Somatic Embryogenesis. International Journal of Molecular Sciences, 24(13), 11077. https://doi.org/10.3390/ijms241311077