

Host–Bacterium Interaction Mechanisms in Staphylococcus aureus Endocarditis: A Systematic Review
Abstract

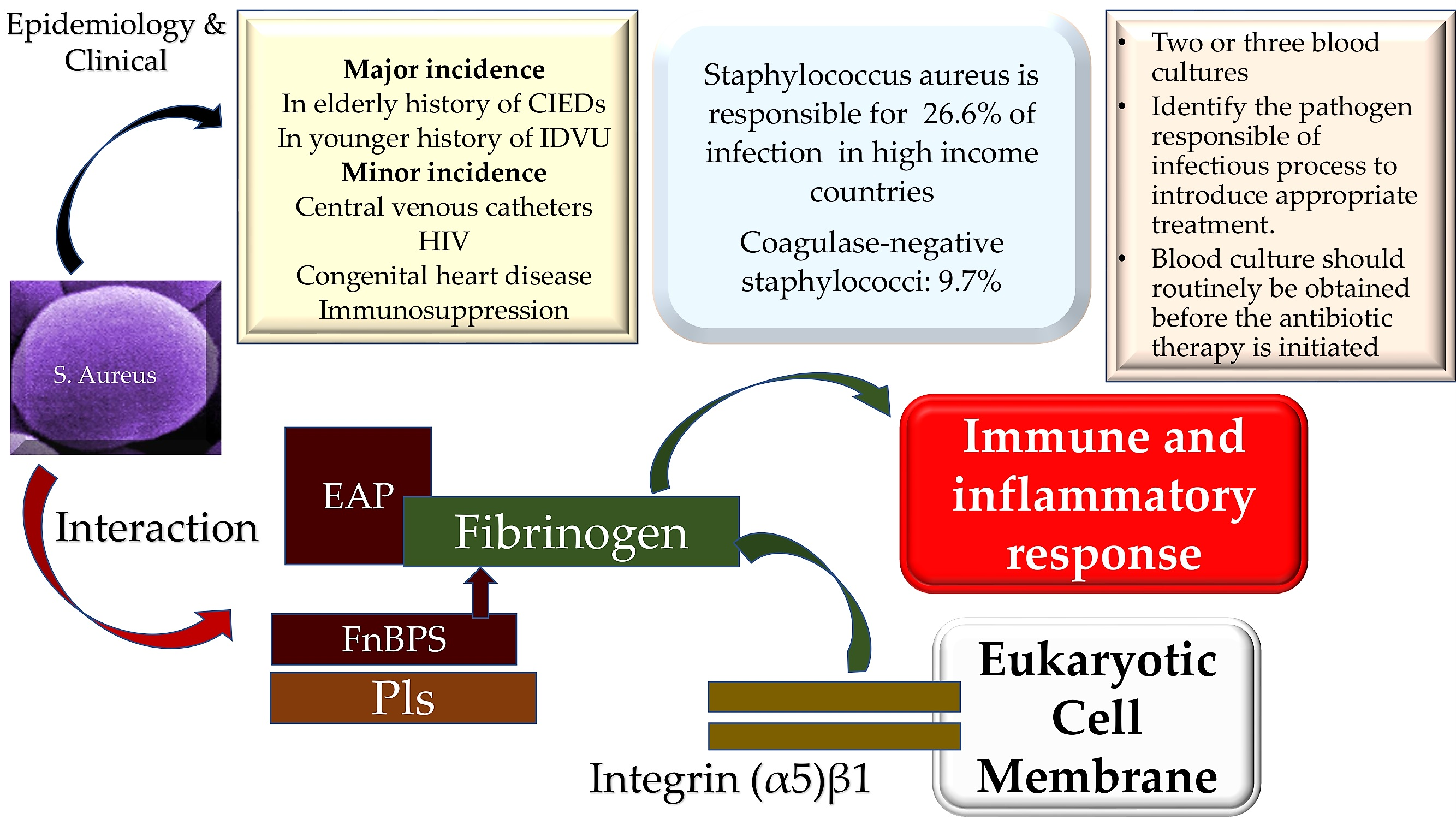
1. Introduction
2. Methods
2.1. Search Strategy
2.2. Study Selection and Data Extraction
2.3. Endpoints and Effect Summary
3. Results and Discussion
3.1. Staphylococcal Manipulation of Host Immune Responses
3.2. Subversion of Innate Immune Responses
3.3. Host–Bacterium Interaction Mechanisms in Staphylococcus aureus Endocarditis
3.4. Immuno-Response and Vaccine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author/Year/Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Lockhart et al. (2008) Circulation [46] | Human RCT Single Center (USA) | 290 pts Brushing Gro 98 vs. Extraction-Amoxicillin 96 vs. Extraction-Placebo 96 | To compare the incidence, duration, nature, and magnitude of IE-related bacteremia from single-tooth extraction and toothbrushing. To determine the impact of amoxicillin prophylaxis on single-tooth extraction. | Amoxicillin has a significant impact on bacteremia resulting from a single-tooth extraction. Toothbrushing may be a greater threat for individuals at risk for infective endocarditis. |
| Mancini et al. (2018) Virulence [49] | Animal (Switzerland pilot) | Rat with catheter-induced aortic vegetations | To investigate the role of Coa and vWbp in IE initiation | Coa does not support the initial colonization of IE (in L. lactis). vWbp contributes to the initiation of IE (in L. lactis) however is marginal in the presence of ClfA. |
| Reguiero et al. (2019) Circ. Cardiovasc. Interv. [51] | Human Comparative Multicenter (Canada pilot) | 245 pts SEV 115 vs. BEV 130 | To determine the incidence, clinical characteristics, and outcomes of patients with IE post-TAVR | IE post-TAVR did not reveal early or late mortality |
| Rodríguez-Vidigal et al. (2019) Enferm. Infecc. Microbiol. Clin. [52] | Human Observational Retrospective (Spain) | 200 pts with TAVI | To evaluate single-centre experience of incidence, mortality, and associated factors of IE after TAVI. | Incidence of IE post-TAVI greater than other series. |
| Di Carluccio et al. (2021) RSC Chem. Biol. [55] | Human Multicenter (Italy pilot) | Collected anatomical specimen | To evaluate the mechanism of interaction of SLBR-B and SLBR-H from S. gordonii in causing IE | Streptococcal Siglec-like adhesins spark the development of tailored synthetic inhibitors and therapeutics specific for Streptococcal adhesins to counteract IE. No impairment of the interplay between Siglecs and glycans. |
| Manukumar et al. (2017) Sci. Rep. [56] | Human Single Center (India) | Collected blood draws | To characterize MRSA strain using MALDI-Biotyper multiplex PCR to distinguish between MRSA and MSSA. To screen PCR-SSCP | PCR-SSCP technique for rapid detection of MSSA and MRSA strains was developed |
| Mempel et al. (2002) Br. J. Dermatol. [57] | Human Single Center (Germany) | † S. aureus DU 5720 vs. S. aureus DU 8325-4 vs. S. aureus DU 5883 | To investigate haemolysin-independent virulence in human keratinocytes. | Staphylococcal invasion of human keratinocytes independently of alpha- and beta-hemolysins, leads to necrotic and apoptotic cell damage. |
| Nakagawa et al. (2017) Cell Host Microbe J. [58] | Animal Multicenter Center (Japan pilot) | Murine epicutaneous infection model | To evaluate how S. aureus trigger inflammation | Increased production of IL-1α, IL-36α and Il 17 via IL-1R and IL-36R. Increased γδ T cells, ILC3 and neutrophil. Keratinocyte * Myd88 signaling in response to S. aureus PSMα drives an IL-17-mediated skin inflammatory response to epicutaneous S. aureus infection. |
| Schwarz et al. (2021) Virulence [63] | Human in vitro and in vivo Multicenter (Germany) | 34 S. aureus Pts with S. aureus endocarditis vs. healthy individuals | To evaluate pathomechanisms in the induction of IE | in vitro assays did not correlate with the severity of IE. S. aureus isolates differed in the activation and inhibition of pathways connected to the extracellular matrix and inflammatory response |
| Malachowa et al. (2011) PLoS ONE [64] | Human/Animal Single center (USA) | S. aureus LAC vs. S. aureus LACΔhlgABC | To study the S. aureus USA300 transcriptome | Limited contribution of any single two-component leukotoxin lukS-PV and lukF-PV to USA300 immune evasion and virulence. |
| Alonso et al. (2013) Nature [65] | Animal Single center (USA) | CCR5-deficient mice | To study activity of S. aureus leukotoxin ED (LukED) | CCR5-deficient mice are resistant to lethal S. aureus infection |
| Kim et al. (2010) J. Exp. Med. [71] | Animal Single center (USA) | λ Mice with SpA (KKAA) | To study S. aureus protective immunity. | SpA (KKAA) immunization enabled MRSA-challenged mice to organize antibody responses to many different staphylococcal antigens. |
| Becker et al. (2014) Proc. Natl. Acad. Sci. USA [72] | In vitro Single center (USA) | S. aureus Newman cultures | To demonstrate that SpA is released with murein tetrapeptide-tetraglycyl [L-Ala-D-iGln-(SpA-Gly5) L-Lys-D-Ala-Gly4] linked to its C-terminal threonyl | SpA, a B cell superantigen, is released with peptidoglycan linked to its C terminus. Murein hydrolases cleave the anchor structure of released SpA to modify host immune responses. |
| Zhang et al. (2015) Infect. Immun. [84] | Animal Single center (China) | Mice SaEsxA and SaEsxB vs. Mice rSaEsxA and rSaEsxB | To investigate SaEsxA and SaEsxB, as possible targets for a vaccine. | SaEsxA and SaEsxB are effective toward Th1 and Th17 candidate antigens. |
| Brady et al. (2013) PLoS ONE [85] | Animal Single center (USA) | Mice HlaH35L vs. Control vs. Prosthetic implant model of chronic biofilm | To evaluate the ability of one S. aureus vaccine antigen to protect in three mouse models of infection | Vaccines may confer protection against one form of S. aureus disease without conferring protection against other disease presentations |
| Zhang et al. (2018) mBio [86] | Animal Multicenter (USA pilot) | C57BL/6 mice | To study the role of adaptive immunity induced by an S. aureus vaccine in protection against S. aureus bacteremia | Multipronged humoral and cellular (B-cell, Th1, Th17) responses to S. aureus antigens may be critical to achieve effective and comprehensive immune defense |
| Yu et al. (2018) Sci. Rep. [87] | Animal Single center (China) | Mouse peritonitis model | To evaluate the humoral immune response and CD4+ T cell-mediated immune responses | The MntC-specific antibodies and MntC-specific Th17 cells play cooperative roles in the prevention of S. aureus infection. |
3.5. Biofilm Formation
| First Author/Year/Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Schwartz et al. (2021) APMIS [88] | In vitro patch enriched with platelet and Leucocyte-rich fibrin Multicenter (Danemark) | IE organoid-like model by colonization with IE-associated bacterial isolates S. aureus, S. mitis and Enterococcus faecalis (IE vegetation (IEV) | To establish an in vitro vegetation simulation IE model for fast screening of novel treatment strategies | The surface-associated bacteria displayed increased tolerance to antibiotics compared to planktonic bacteria. IE simulation model with the relevant pathogens S. aureus, S. mitis group, and E. faecalis was established and IE model mirrors the natural IE process |
| Di Domenico et al. (2019) BMC Microbiol. [89] | Human Multicenter (IT) | Samples of infected heart tissue. S. aureus 50%, Enterococcus faecalis 25% and Streptococcus gallolyticus 25% | To assess a rapid biofilm identification assay and a targeted antimicrobial susceptibility profile of biofilm-growing bacteria in patients with IE, which were unresponsive to antibiotic therapy | Biofilm-producing bacteria, from surgically treated IE, display a high tolerance to antibiotics, which is undetected by conventional antibiograms |
| Schwartz et al. (2012) APMIS [90] | Animal model Multicenter (Danemark) | IE organoid-like model by colonization with IE-associated bacterial isolates S. aureus, S. mitis and Enterococcus faecalis (IEV) | To evaluate the time course of biofilm formation and the impact on antibiotic tolerance development | The antibiotic effect was significantly higher than when treatment was started after the biofilm was allowed to mature |
| Kim et al. (2016) JTCVS [95] | Human Single Center (USA) | 86 pts Homografts vs. 139 pts Xenograft prostheses vs. 79 pts Mechanical prostheses | To evaluate resistance to infection | Homografts were more used in PVE (p = 0.002) and methicillin-resistant Staphylococcus (p = 0.002), compared with conventional prostheses. No significant benefit to the use of homografts was demonstrable with regard to resistance to reinfection in the setting of IE |
| Nappi et al. (2018) JTCVS [102] | Human Single center (France) | 210 pts | To evaluate long-term results of aortic allografts and to identify factors influencing long-term durability | The use of allograft is a valid option in complex infective endocarditis and in women of childbearing age |
| Steffen et al. (2016) JTCVS [113] | In vitro Single center (Germany) | 10 cryopreserved human allografts | To evaluate the in vitro antimicrobial activity of 3 antibiotic regimens | Allograft antibacterial activity despite long-term storage over 5 years. Antibiotic combinations applied during CHA processing have a significant influence on their infection resistance. Ascending aortic tissue shows a significantly enhanced bacterial resistance against staphylococcal bacteria compared with aortic valves |
3.6. Interaction of Staphylococcus aureus with Coagulation Mechanisms
3.7. Involvement of Vascular Endothelium and Blood Constituents in S. aureus-Induced Endocarditis
3.8. Infective Endocarditis and Platelets
| First Author/Year/Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Que et al. (2005) J. Exp. Med. [73] | Animal model Single Center (Switzerland) | Rat model of IE induced | To study valve colonization with experimental endocarditis. To evaluate the role of ClfA and FnBPA positive lactococci | Fibrinogen and fibronectin binding could cooperate for S. aureus valve colonization and endothelial invasion in vivo |
| Edwards et al. (2012) PLoS ONE [74] | Human Single Center (UK) | Blood sample | To study in vivo role of Eap to interact with host glyco-proteins | Eap expressing strains cause a more severe infection, demonstrating its role in invasive disease. Increased level of TNFα and gC1qR/p33 expression |
| Veloso et al. (2013) Infect. Immun. [76] | Animal model Single Center (Switzerland) | Rat model of induced IE 10(6) CFU L. lactis pIL253 vs. Recombinant L. lactis (ClfA, FnbpA, BCD, or SdrE) | To explore the contributions of S. aureus virulence factors to the initiation of IE | Fibrinogen binding in the initiation of S. aureus IE. Activation of platelet aggregation or an inflammatory response may contribute to or promote the development of EI |
| Thomas et al. (2021) mBio [77] | Animal model Single Center (USA) | Rat model of IE induced | To identify proteins with significant amino acid identities to vWbp | Protein homologous to the C-terminal of vWbp was identified. Its role in Fg shield assembly and binds |
| Hussain et al. (2002) Infect. Immun. [78] | In vitro Single center (Germany) | S. aureus Newman cultures vs. Control mutant | To investigate the role of Eap by constructing a stable eap::ermB deletion | Eap may contribute to pathogenicity by promoting adhesion of whole staphylococcal cells to complex eukaryotic substrates |
| Palankar et al. (2018) Int. J. Med. Microbiol. [79] | In vitro Single center (Germany) | S. aureus Mu50 | To investigate Eap subdomain and interaction with platelet | Eap subdomain Eap D3D4 specifically interacts and rapidly activates human platelets |
| Hussain et al. (2008) Infect. Immun. [80] | In vitro Single center (Germany) | S. aureus Newman cultures vs. S. aureus Wood 46 | To investigate the interactions of full-length Eap and five recombinant tandem repeat domains with host proteins | More than one Eap tandem repeat domain is required for S. aureus agglutination, adherence, and cellular invasion but not for the stimulation of PBMC proliferation |
| Heying et al. (2007) Thromb. Haemost. [114] | Human Single Center (Germany) | S. aureus L. lactis culture cultured human EC | To investigate the role of FnBPA, FnBPB ClfA to promote bacterial adherence to cultured human ECs | S. aureus FnBPs, but not ClfA, lead pathogenicity to non-pathogenic L. lactis. Adhesins (ICAM-1 and VCAM-1) evokes inflammation (interleukin-8) as well as procoagulant activity |
| Piroth et al. (2008) Infect. Immun. [115] | Animal model Single Center (Switzerland) | S. aureus L. lactis culture In vitro and in vivo | To study the subdomain of FnBPA responsible for fibrinogen and fibronectin binding, cell invasion, and in vivo endocarditis | Fb binding combined with fibronectin binding to synergize the invasion of cultured cell lines is correlated with IE severity |
| Pappelbaum et al. (2013) Circulation [117] | Human/Animal Single center (Germany) | 6 WT mice with VWF vs. 5 knockout mice vs. Cultured human EC | Whether ULVWF mediates bacterial adherence | ULVWF contributes to the initial pathogenic step of S aureus-induced endocarditis in patients with an intact endothelium. Heparin or ADAMTS13 intervenes in decreasing ULVWF adherence |
| Claes et al. (2018) Thromb. Haemost. [119] | Human/Animal Multicenter (Belgium pilot) | L. lactis-clfA vs. L. lactis-fnbpA vs. Cultured human EC | To study the influence of shear flow and plasma on the binding of ClfA and FnbpA | Pharmacological inhibition of ClfA-Fg interactions may constitute a valuable additive treatment in infective endocarditis |
| Ko et al. (2016) mBio [120] | Animal model Single Center (USA) | Rat model of IE induced | To identify variants of a linear Fg binding motif, present in Coa and Efb which are responsible for the Fg binding activities of these proteins | S. aureus coagulase can induce the formation of a fibrinogen shield in experimental abscess models which surrounds and protects bacteria in the microcolony from clearance |
| Niemann et al. (2021) mBio [125] | Animal Multicenter (Germany) | Rat model of IE induced in osteoblasts vs. epithelial cells | To demonstrate that S. aureus was less engulfed in osteoblasts than in epithelial cells | Large differences of S. aureus uptake efficacy in different host cell types. In vivo differences between courses of bacterial infections and the localization of bacteria in different clinical settings mediated by α5β1-integrin |
| Pietrocola et al. (2020) J. Biol. Chem. [126] | Animal Multicenter center (Italy pilot) | Rat model of IE induced | To evaluate a variety of virulence factors that promote infection by S. aureus | Adherence to and invasion of epithelial and ECs by IsdB-expressing S. aureus cells was promoted by Vn, and an αvβ3 integrin-blocking mAb |
| Alfeo et al. (2021) Sci. Rep. [127] | Animal Multicenter center (Italy pilot) | Rat model of IE induced | To study IsdB protein and Vn binding Interacts with vWF | Importance of IsdB in adherence of S. aureus to the endothelium colonization and as potential therapeutic target |
| Ditkowski et al. (2021) J. Thorac. Cardiovasc. Surg. [129] | Human Multicenter (Belgium pilot) | 5 graft tissues | To investigate contributions by platelets and plasma fibrinogen to IE initiation on various grafts used for valve replacement | Binding of plasma Fg to especially BJV grafts enables adhesion of single platelets via αIIbβ3. S aureus attaches from blood to activated bound platelet αIIbβ3 via plasma fibrinogen |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selton-Suty, C.; Celard, M.; Le Moing, V. Preeminence of Staphylococcus aureus in infective endocarditis: A 1-year population-based survey. Clin. Infect. Dis. 2012, 54, 1230–1239. [Google Scholar] [CrossRef]
- Chen, H.; Zhan, Y.; Zhang, K.; Gao, Y.; Chen, L.; Zhan, J.; Chen, Z.; Zeng, Z. The Global, Regional, and National Burden and Trends of Infective Endocarditis From 1990 to 2019: Results from the Global Burden of Disease Study 2019. Front. Med. 2022, 9, 774224. [Google Scholar] [CrossRef] [PubMed]
- Resende, P., Jr.; Fortes, C.Q.; do Nascimento, E.M.; Sousa, C.; Querido Fortes, N.R.; Thomaz, D.C.; de Bragança Pereira, B.; Pinto, F.J.; de Oliveira, G.M.M. In-hospital Outcomes of Infective Endocarditis from 1978 to 2015: Analysis Through Machine-Learning Techniques. CJC Open 2021, 4, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Allegranzi, B.; Bagheri Nejad, S.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of endemic health-care-associated infection in developing countries: Systematic review and meta-analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Bagheri Nejad, S.; Allegranzi, B.; Syed, S.B.; Ellis, B.; Pittet, D. Health-care-associated infection in Africa: A systematic review. Bull. World Health Organ. 2011, 89, 757–765. [Google Scholar] [CrossRef]
- Joubert, D.; Cullati, S.; Briot, P.; Righi, L.; Grauser, D.; Ourahmoune, A.; Chopard, P. How to improve hospital admission screening for patients at risk of multidrug-resistant organism carriage: A before-and-after interventional study and cost-effectiveness analysis. BMJ Open Qual. 2022, 11, e001699. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L. Antibiotics and antibiotic resistance genes in natural environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, J.; Wei, Y.; Zhang, J.; Tao, C. Recent advances in metal-organic framework-based materials for anti-staphylococcus aureus infection. Nano Res. 2022, 11, 6220–6242. [Google Scholar] [CrossRef]
- McAdow, M.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus secretes coagulase and von Willebrand factor binding protein to modify the coagulation cascade and establish host infections. J. Innate Immun. 2012, 4, 141–148. [Google Scholar] [CrossRef]
- Thomer, L.; Schneewind, O.; Missiakas, D. Multiple ligands of von Willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J. Biol. Chem. 2013, 288, 28283–28292. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Martuscelli, G.; Bellomo, F.; Avtaar Singh, S.S.; Moon, M.R. Infective Endocarditis in High-Income Countries. Metabolites 2022, 12, 682. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Liu, W.; Arora, S.; Ganesh, V.; Ko, Y.P.; Höök, M. The Complex Fibrinogen Interactions of the Staphylococcus aureus Coagulases. Front. Cell. Infect. Microbiol. 2019, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Herrmann, M. Mechanism and consequences of invasion of endothelial cells by Staphylococcus aureus. Thromb. Haemost. 2005, 94, 266–277. [Google Scholar]
- Thuny, F.; Di Salvo, G.; Belliard, O.; Avierinos, J.F.; Pergola, V.; Rosenberg, V.; Casalta, J.P.; Gouvernet, J.; Derumeaux, G.; Iarussi, D.; et al. Risk of embolism and death in infective endocarditis: Prognostic value of echocardiography: A prospective multicenter study. Circulation 2005, 112, 69–75, Erratum in Circulation 2005, 112, e125. [Google Scholar] [CrossRef]
- Di Salvo, G.; Habib, G.; Pergola, V.; Avierinos, J.F.; Philip, E.; Casalta, J.P.; Vailloud, J.M.; Derumeaux, G.; Gouvernet, J.; Ambrosi, P.; et al. Echocardiography predicts embolic events in infective endocarditis. J. Am. Coll. Cardiol. 2001, 37, 1069–1076. [Google Scholar] [CrossRef]
- Vilacosta, I.; Graupner, C.; San Román, J.A.; Sarriá, C.; Ronderos, R.; Fernández, C.; Mancini, L.; Sanz, O.; Sanmartín, J.V.; Stoermann, W. Risk of embolization after institution of antibiotic therapy for infective endocarditis. J. Am. Coll. Cardiol. 2002, 39, 1489–1495. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Dreyfus, J.; Attias, D.; Acar, C.; Bando, K. Mitral endocarditis: A new management framework. J. Thorac. Cardiovasc. Surg. 2018, 156, 1486–1495.e4. [Google Scholar] [CrossRef]
- Avtaar Singh, S.S.; Costantino, M.F.; D’Addeo, G.; Cardinale, D.; Fiorilli, R.; Nappi, F. A narrative review of diagnosis of infective endocarditis-imaging methods and comparison. Ann. Transl. Med. 2020, 8, 1621. [Google Scholar] [CrossRef]
- Duval, X.; Iung, B.; Klein, I.; Brochet, E.; Thabut, G.; Arnoult, F.; Lepage, L.; Laissy, J.P.; Wolff, M.; Leport, C.; et al. Effect of early cerebral magnetic resonance imaging on clinical decisions in infective endocarditis: A prospective study. Ann. Intern. Med. 2010, 152, 497–504. [Google Scholar] [CrossRef]
- Béraud, G.; Tubiana, S.; Erpelding, M.L.; Le Moing, V.; Chirouze, C.; Gorenne, I.; Manchon, P.; Tattevin, P.; Vernet, V.; Varon, E.; et al. Combined Bacterial Meningitis and Infective Endocarditis: When Should We Search for the Other When Either One is Diagnosed? Infect. Dis. Ther. 2022, 11, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Vitali, P.; Savoldi, F.; Segati, F.; Melazzini, L.; Zanardo, M.; Fedeli, M.P.; Benedek, A.; Di Leo, G.; Menicanti, L.; Sardanelli, F. MRI versus CT in the detection of brain lesions in patients with infective endocarditis before or after cardiac surgery. Neuroradiology 2022, 64, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Corr, P.; Wright, M.; Handler, L.C. Endocarditis- related cerebral aneurysms: Radiologic changes with treatment. AJNR Am. J. Neuroradiol. 1995, 16, 745–748. [Google Scholar] [PubMed]
- Champey, J.; Pavese, P.; Bouvaist, H.; Maillet, M.; Kastler, A.; Boussat, B.; Francois, P.; The Investigator Groups. Is brain angio-MRI useful in infective endocarditis management? Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 2053–2058. [Google Scholar] [CrossRef]
- Peters, P.J.; Harrison, T.; Lennox, J.L. A dangerous dilemma: Management of infectious intracranial aneurysms complicating endocarditis. Lancet Infect. Dis. 2006, 6, 742–748. [Google Scholar] [CrossRef]
- Serrano, F.; Guédon, A.; Saint-Maurice, J.P.; Labeyrie, M.A.; Civelli, V.; Eliezer, M.; Houdart, E. Endovascular treatment of infectious intracranial aneurysms complicating infective endocarditis: A series of 31 patients with 55 aneurysms. Neuroradiology 2022, 64, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, D.R.; Corey, G.R.; Hoen, B.; Miró, J.M.; Fowler VGJr Bayer, A.S.; Karchmer, A.W.; Olaison, L.; Pappas, P.A.; Moreillon, P.; Chambers, S.T.; et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: The International Collaboration on Endocarditis-Prospective Cohort Study. Arch. Intern. Med. 2009, 169, 463–473. [Google Scholar] [CrossRef]
- Liaqat, W.; Palaiodimos, L.; Li, W.; Karamanis, D.; Tahir, A.; Tzoumas, A.; Nagraj, S.; Tiwari, N.; Grushko, M.; Kokkinidis, D.; et al. Epidemiologic and clinical characteristics of infective endocarditis: A single-center retrospective study in the Bronx, New York. Infection 2022, 50, 1349–1361. [Google Scholar] [CrossRef]
- Paul, G.; Ochs, L.; Hohmann, C.; Baldus, S.; Michels, G.; Meyer-Schwickerath, C.; Fätkenheuer, G.; Mader, N.; Wahlers, T.; Weber, C.; et al. Surgical Procedure Time and Mortality in Patients with Infective Endocarditis Caused by Staphylococcus aureus or Streptococcus Species. J. Clin. Med. 2022, 11, 2538. [Google Scholar] [CrossRef]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef]
- López, J.; Revilla, A.; Vilacosta, I.; Villacorta, E.; González-Juanatey, C.; Gómez, I.; Rollán, M.J.; San Román, J.A. Definition, clinical profile, microbiological spectrum, and prognostic factors of early-onset prosthetic valve endocarditis. Eur. Heart J. 2007, 28, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Valle, H.; Fariñas-Alvarez, C.; García-Palomo, J.D.; Bernal, J.M.; Martín-Durán, R.; Gutiérrez Díez, J.F.; Revuelta, J.M.; Fariñas, M.C. Clinical course and predictors of death in prosthetic valve endocarditis over a 20-year period. J. Thorac. Cardiovasc. Surg. 2010, 139, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Chen, X.; Tang, A.; Huang, D.; Pan, Q.; Fang, Z. Prevalence and Molecular Characterization of Methicillin-Resistant Staphylococci Recovered from Public Shared Bicycles in China. Int. J. Environ. Res. Public Health 2022, 19, 4492. [Google Scholar] [CrossRef] [PubMed]
- Argemi, X.; Hansmann, Y.; Prola, K.; Prévost, G. Coagulase-Negative Staphylococci Pathogenomics. Int. J. Mol. Sci. 2019, 20, 1215. [Google Scholar] [CrossRef]
- Chu, V.H.; Woods, C.W.; Miro, J.M.; Cabell, C.H.; Pappas, P.A.; Federspiel, J.; Athan, E.; Stryjewski, M.E.; Nacinovich, F.; Marco, F.; et al. Emergence of coagulase-negative staphylococci as a cause of native valve endocarditis. Clin. Infect. Dis. 2008, 46, 232–242. [Google Scholar] [CrossRef]
- Chu, V.H.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Pappas, P.A.; Jones, P.; Stryjewski, M.E.; Anguera, I.; Braun, S.; Muñoz, P.; et al. International Collaboration on Endocarditis-Prospective Cohort Study Group. Coagulase-negative staphylococcal prosthetic valve endocarditis—A contemporary update based on the International Collaboration on Endocarditis: Prospective cohort study. Heart 2009, 95, 570–576. [Google Scholar] [CrossRef]
- Alawad, M.J.; Ali, G.A.; Goravey, W. Underrecognized pathogen; Staphylococcus warneri-associated native mitral valve endocarditis in an immunocompetent host: A case report and literature review. Clin. Case Rep. 2022, 10, e05591. [Google Scholar] [CrossRef]
- Voigt, A.; Shalaby, A.; Saba, S. Rising rates of cardiac rhythm management device infections in the United States: 1996 through 2003. J. Am. Coll. Cardiol. 2006, 48, 590–591. [Google Scholar] [CrossRef]
- Traykov, V.; Blomström-Lundqvist, C. Antibiotic-Eluting Envelopes for the Prevention of Cardiac Implantable Electronic Device Infections: Rationale, Efficacy, and Cost-Effectiveness. Front. Cardiovasc. Med. 2022, 9, 855233. [Google Scholar] [CrossRef]
- Elad, B.; Perl, L.; Hamdan, A.; Yahav, D.; Atamna, A.; Shaked, H.; Rubchevsky, V.; Sharony, R.; Bernstine, H.; Shapira, Y.; et al. The clinical value of the endocarditis team: Insights from before and after guidelines implementation strategy. Infection 2022, 50, 57–64. [Google Scholar] [CrossRef]
- Han, H.C.; Hawkins, N.M.; Pearman, C.M.; Birnie, D.H.; Krahn, A.D. Epidemiology of cardiac implantable electronic device infections: Incidence and risk factors. Europace 2021, 23 (Suppl. S4), iv3–iv10. [Google Scholar] [CrossRef] [PubMed]
- Durante-Mangoni, E.; Bradley, S.; Selton-Suty, C.; Tripodi, M.F.; Barsic, B.; Bouza, E.; Cabell, C.H.; Ramos, A.I.; Fowler VJr Hoen, B.; Koneçny, P.; et al. Current features of infective endocarditis in elderly patients: Results of the International Collaboration on Endocarditis Prospective Cohort Study. Arch. Intern. Med. 2008, 168, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Iossa, D.; Ursi, M.P.; Bertolino, L.; Karruli, A.; Molaro, R.; Esposito, G.; Vitrone, M.; D’Amico, F.; Albisinni, R.; et al. Clinical Significance and Prognostic Value of Hemostasis Parameters in 337 Patients with Acute Infective Endocarditis. J. Clin. Med. 2021, 10, 5386. [Google Scholar] [CrossRef] [PubMed]
- Molton, J.S.; Tambyah, P.A.; Ang, B.S.P.; Ling, M.L.; Fisher, D.A. The global spread of healthcare-associated multidrug-resistant bacteria: A perspective from Asia. Clin. Infect. Dis. 2013, 56, 1310–1318. [Google Scholar]
- Çaǧlayan, Ç.; Barnes, S.L.; Pineles, L.L.; Harris, A.D.; Klein, E.Y. A Data-Driven Framework for Identifying Intensive Care Unit Admissions Colonized With Multidrug-Resistant Organisms. Front. Public Health 2022, 10, 853757. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef]
- Widmer, E.; Que, Y.A.; Entenza, J.M.; Moreillon, P. New concepts in the pathophysiology of infective endocarditis. Curr. Infect. Dis. Rep. 2006, 8, 271–279. [Google Scholar] [CrossRef]
- Moreillon, P.; Que, Y.A.; Bayer, A.S. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect. Dis. Clin. N. Am. 2002, 16, 297–318. [Google Scholar] [CrossRef]
- Mancini, S.; Oechslin, F.; Menzi, C.; Que, Y.A.; Claes, J.; Heying, R.; Veloso, T.R.; Vanassche, T.; Missiakas, D.; Schneewind, O.; et al. Marginal role of von Willebrand factor-binding protein and coagulase in the initiation of endocarditis in rats with catheter-induced aortic vegetations. Virulence 2018, 9, 1615–1624. [Google Scholar] [CrossRef]
- Werdan, K.; Dietz, S.; Löffl, B.; Niemann, S.; Bushnaq, H.; Silber, R.E.; Peters, G.; Müller-Werdan, U. Mechanisms of infective Endocarditis: Pathogen-host interaction and risk states. Nat. Rev. Cardiol. 2014, 11, 35–50. [Google Scholar] [CrossRef]
- Regueiro, A.; Linke, A.; Latib, A.; Ihlemann, N.; Urena, M.; Walther, T.; Husser, O.; Herrmann, C.; Nombela-Franco, L.; Cheema, A.; et al. Infective Endocarditis Following Transcatheter Aortic Valve Replacement: Comparison of Balloon- Versus Self-Expandable Valves. Circ. Cardiovasc. Interv. 2019, 12, e007938. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vidigal, F.F.; Nogales-Asensio, J.M.; Calvo-Cano, A.; González-Fernández, R.; Martínez-Carapeto, A.; Gómez-Sanchez, I.; Bengla-Limpo, B.; Merchán-Herrera, A.; Nogales-Muñoz, N.; Vera-Tomé, A.; et al. Infective endocarditis after transcatheter aortic valve implantation: Contributions of a single-centre experience on incidence and associated factors. Enferm. Infecc. Microbiol. Clin. (Engl. Ed). 2019, 37, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Ciofu, O.; Moser, C.; Jensen, P.Ø.; Høiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 2022, 20, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Annappah, D.; Saling, M.; Prodafikas, J.; Badie, A.N. Device-associated aortic valve endocarditis due to a complicated Enterobacter cloacae urinary tract infection. ID Cases 2021, 27, e01365. [Google Scholar] [CrossRef] [PubMed]
- Di Carluccio, C.; Forgione, R.E.; Bosso, A.; Yokoyama, S.; Manabe, Y.; Pizzo, E.; Molinaro, A.; Fukase, K.; Fragai, M.; Bensing, B.A.; et al. Molecular recognition of sialoglycans by streptococcal Siglec-like adhesins: Toward the shape of specific inhibitors. RSC Chem. Biol. 2021, 2, 1618–1630. [Google Scholar] [CrossRef]
- Manukumar, H.M.; Umesha, S. MALDI-TOF-MS based identification and molecular characterization of food associated methicillin-resistant Staphylococcus aureus. Sci. Rep. 2017, 7, 11414. [Google Scholar] [CrossRef]
- Mempel, M.; Schnopp, C.; Hojka, M.; Fesq, H.; Weidinger, S.; Schaller, M.; Korting, H.C.; Ring, J.; Abeck, D. Invasion of human keratinocytes by Staphylococcus aureus and intracellular bacterial persistence represent haemolysin-independent virulence mechanisms that are followed by features of necrotic and apoptotic keratinocyte cell death. Br. J. Dermatol. 2002, 146, 943–951. [Google Scholar] [CrossRef]
- Nakagawa, S.; Matsumoto, M.; Katayama, Y.; Oguma, R.; Wakabayashi, S.; Nygaard, T.; Saijo, S.; Inohara, N.; Otto, M.; Matsue, H.; et al. Staphylococcus aureus virulent PSMα peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe 2017, 22, 667–677.e5. [Google Scholar] [CrossRef]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef]
- Kawa, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kupper, T.S.; Fuhlbrigge, R.C. Immune surveillance in the skin: Mechanisms and clinical consequences. Nat. Rev. Immunol. 2004, 4, 211–222. [Google Scholar] [CrossRef]
- Nestle, F.O.; Di, M.P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Töre, Y.; Hoesker, V.; Ameling, S.; Grün, K.; Völker, U.; Schulze, P.C.; Franz, M.; Faber, C.; Schaumburg, F.; et al. Host-pathogen interactions of clinical S. aureus isolates to induce infective endocarditis. Virulence 2021, 12, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS ONE 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., 3rd; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F., 3rd; Torres, V.J. Bacterial survival amidst an immune onslaught: The contribution of the Staphylococcus aureus leukotoxins. PLoS Pathog. 2013, 9, e1003143. [Google Scholar] [CrossRef]
- Cheung, G.Y.; Joo, H.S.; Chatterjee, S.S.; Otto, M. Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol. Rev. 2014, 38, 698–719. [Google Scholar] [CrossRef]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef]
- Silverman, G.J.; Goodyear, C.S. Confounding B-cell defences: Lessons from a staphylococcal superantigen. Nat. Rev. Immunol. 2006, 6, 465–475. [Google Scholar] [CrossRef]
- Kim, H.K.; Cheng, A.G.; Kim, H.Y.; Missiakas, D.M.; Schneewind, O. Nontoxigenic protein A vaccine for methicillin-resistant Staphylococcus aureus infections in mice. J. Exp. Med. 2010, 207, 1863–1870. [Google Scholar] [CrossRef]
- Becker, S.; Frankel, M.B.; Schneewind, O.; Missiakas, D. Release of protein A from the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2014, 111, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.-A.; Haefliger, J.-A.; Piroth, L.; François, P.; Widmer, E.; Entenza, J.M.; Sinha, B.; Herrmann, M.; Francioli, P.; Vaudaux, P.; et al. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J. Exp. Med. 2005, 201, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.M.; Bowden, M.G.; Brown, E.L.; Laabei, M.; Massey, R.C. Staphylococcus aureus extracellular adherence protein triggers TNFα release, promoting attachment to endothelial cells via protein A. PLoS ONE 2012, 7, e43046. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat. Rev. Microbiol. 2006, 4, 445–457. [Google Scholar] [CrossRef]
- Veloso, T.R.; Chaouch, A.; Roger, T.; Giddey, M.; Vouillamoz, J.; Majcherczyk, P.; Que, Y.A.; Rousson, V.; Moreillon, P.; Entenza, J.M. Use of a human-like low-grade bacteremia model of experimental endocarditis to study the role of Staphylococcus aureus adhesins and platelet aggregation in early endocarditis. Infect. Immun. 2013, 81, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Arora, S.; Liu, W.; Churion, K.; Wu, Y.; Höök, M. vhp Is a Fibrinogen-Binding Protein Related to vWbp in Staphylococcus aureus. mBio 2021, 12, e0116721. [Google Scholar] [CrossRef]
- Hussain, M.; Haggar, A.; Heilmann, C.; Peters, G.; Flock, J.I.; Herrmann, M. Insertional inactivation of Eap in Staphylococcus aureus strain Newman confers reduced staphylococcal binding to fibroblasts. Infect. Immun. 2002, 70, 2933–2940. [Google Scholar] [CrossRef]
- Palankar, R.; Binsker, U.; Haracska, B.; Wesche, J.; Greinacher, A.; Hammerschmidt, S. Interaction between the Staphylococcus aureus extracellular adherence protein Eap and its subdomains with platelets. Int. J. Med. Microbiol. 2018, 308, 683–691. [Google Scholar] [CrossRef]
- Hussain, M.; Haggar, A.; Peters, G.; Chhatwal, G.S.; Herrmann, M.; Flock, J.I.; Sinha, B. More than one tandem repeat domain of the extracellular adherence protein of Staphylococcus aureus is required for aggregation, adherence, and host cell invasion but not for leukocyte activation. Infect. Immun. 2008, 76, 5615–5623. [Google Scholar] [CrossRef]
- Harraghy, N.; Hussain, M.; Haggar, A.; Chavakis, T.; Sinha, B.; Herrmann, M.; Flock, J.I. The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap. Microbiology 2003, 149 Pt 10, 2701–2707. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Zhang, X.; Marichannegowda, M.H.; Rakesh, K.P.; Qin, H.L. Master mechanisms of Staphylococcus aureus: Consider its excellent protective mechanisms hindering vaccine development! Microbiol. Res. 2018, 212–213, 59–66. [Google Scholar] [CrossRef]
- Zhang, B.Z.; Hua, Y.H.; Yu, B.; Lau, C.C.; Cai, J.P.; Zheng, S.Y.; Yam, W.C.; Kao, R.Y.; Sze, K.H.; Zheng, B.J.; et al. Recombinant ESAT-6-like proteins provoke protective immune responses against invasive Staphylococcus aureus disease in a murine model. Infect. Immun. 2015, 83, 339–345. [Google Scholar] [CrossRef]
- Brady, R.A.; Mocca, C.P.; Prabhakara, R.; Plaut, R.D.; Shirtliff, M.E.; Merkel, T.J.; Burns, D. Evaluation of genetically inactivated alpha toxin for protection in multiple mouse models of Staphylococcus aureus infection. PLoS ONE 2013, 8, e63040. [Google Scholar] [CrossRef]
- Zhang, F.; Ledue, O.; Jun, M.; Goulart, C.; Malley, R.; Lu, Y.J. Protection against Staphylococcus aureus Colonization and Infection by B- and T-Cell-Mediated Mechanisms. ASM J. mBio 2018, 9, e01949-18. [Google Scholar] [CrossRef]
- Yu, W.; Yao, D.; Yu, S.; Wang, X.; Li, X.; Wang, M.; Liu, S.; Feng, Z.; Chen, X.; Li, W.; et al. Protective humoral and CD4+ T cellular immune responses of Staphylococcus aureus vaccine MntC in a murine peritonitis model. Sci. Rep. 2018, 8, 3580. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, F.A.; Christophersen, L.; Laulund, A.S.; Lundquist, R.; Lerche, C.; Rude Nielsen, P.; Bundgaard, H.; Høiby, N.; Moser, C. Novel human in vitro vegetation simulation model for infective endocarditis. APMIS 2021, 129, 653–662. [Google Scholar] [CrossRef]
- Di Domenico, E.G.; Rimoldi, S.G.; Cavallo ID’Agosto, G.; Trento, E.; Cagnoni, G.; Palazzin, A.; Pagani, C.; Romeri, F.; De Vecchi, E.; Schiavini, M.; et al. Microbial biofilm correlates with an increased antibiotic tolerance and poor therapeutic outcome in infective endocarditis. BMC Microbiol. 2019, 19, 228. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, F.A.; Nielsen, L.; Struve Andersen, J.; Bock, M.; Christophersen, L.; Sunnerhagen, T.; Lerche, C.J.; Bay, L.; Bundgaard, H.; Høiby, N.; et al. Dynamics of a Staphylococcus aureus infective endocarditis simulation model. APMIS 2022, 130, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Avtaar Singh, S.S.; Timofeeva, I. Learning From Controversy: Contemporary Surgical Management of Aortic Valve Endocarditis. Clin. Med. Insights Cardiol. 2020, 14, 1179546820960729. [Google Scholar] [CrossRef]
- Nappi, F.; Singh, S.S.A.; Nappi, P.; Spadaccio, C.; Nenna, A.; Gentile, F.; Chello, M. Heart Valve Endocarditis. Surg. Technol. Int. 2020, 37, 203–215. [Google Scholar] [PubMed]
- Nappi, F.; Singh, S.S.A.; Spadaccio, C.; Acar, C. Revisiting the guidelines and choice the ideal substitute for aortic valve endocarditis. Ann. Transl. Med. 2020, 8, 952. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Iervolino, A.; Singh, S.S.A. The New Challenge for Heart Endocarditis: From Conventional Prosthesis to New Devices and Platforms for the Treatment of Structural Heart Disease. BioMed Res. Int. 2021, 2021, 7302165. [Google Scholar] [CrossRef]
- Kim, J.B.; Ejiofor, J.I.; Yammine, M.; Camuso, J.M.; Walsh, C.W.; Ando, M.; Melnitchouk, S.I.; Rawn, J.D.; Leacche, M.; MacGillivray, T.E.; et al. Are homografts superior to conventional prosthetic valves in the setting of infective endocarditis involving the aortic valve? J. Thorac. Cardiovasc. Surg. 2016, 151, 1239–1248.e2. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, S.; Jeppsson, A.; Fröjd, V.; Svensson, G. Surgical Treatment of Aortic Prosthetic Valve Endocarditis: A 20-Year Single-Center Experience. Ann. Thorac. Surg. 2016, 101, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- David, T.E.; Gavra, G.; Feindel, C.M.; Regesta, T.; Armstrong, S.; Maganti, M.D. Surgical treatment of active infective endocarditis: A continued challenge. J. Thorac. Cardiovasc. Surg. 2007, 133, 144–149. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Acar, C. Use of allogeneic tissue to treat infective valvular disease: Has. everything been said? J. Thorac. Cardiovasc. Surg. 2017, 153, 824–828. [Google Scholar] [CrossRef]
- Brouqui, P.; Raoult, D. Endocarditis due to rare and fastidious bacteria. Clin. Microbiol. Rev. 2001, 14, 177–207. [Google Scholar] [CrossRef]
- Kim, J.B.; Ejiofor, J.I.; Yammine, M.; Ando, M.; Camuso, J.M.; Youngster, I.; Nelson, S.B.; Kim, A.Y.; Melnitchouk, S.I.; Rawn, J.D.; et al. Surgical outcomes of infective endocarditis among intravenous drug users. J. Thorac. Cardiovasc. Surg. 2016, 152, 832–841.e1. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C. Simplest solutions are not always the cleverest: Can. we stitch in an infected annulus? Should we rethink the current guidelines? J. Thorac. Cardiovasc. Surg. 2017, 154, 1899–1900. [Google Scholar] [CrossRef]
- Nappi, F.; Nenna, A.; Petitti, T.; Spadaccio, C.; Gambardella, I.; Lusini, M.; Chello, M.; Acar, C. Long-term outcome of cryopreserved allograft for aortic valve replacement. J. Thorac. Cardiovasc. Surg. 2018, 156, 1357–1365.e6. [Google Scholar] [CrossRef] [PubMed]
- Arabkhani, B.; Bekkers, J.A.; Andrinopoulou, E.-R.; Roos-Hesselink, J.W.; Takkenberg, J.J.M.; Bogers, A.J.J.C. Allografts in aortic position: Insights from a 27-year, single-center prospective study. J. Thorac. Cardiovasc. Surg. 2016, 152, 1572–1579.e3. [Google Scholar] [CrossRef]
- Nenna, A.; Nappi, F.; Dougal, J.; Satriano, U.; Chello, C.; Mastroianni, C.; Lusini, M.; Chello, M.; Spadaccio, C. Sternal wound closure in the current era: The need of a tailored approach. Gen. Thorac. Cardiovasc. Surg. 2019, 67, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, S.; Tesar, P.J.; Pearse, B.; Jalali, H.; Sparks, L.; Fraser, J.F.; Pohlner, P.G. Long-term clinical outcomes after aortic valve replacement using cryopreserved aortic allograft. J. Thorac. Cardiovasc. Surg. 2014, 148, 65–72.e2. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.F.; Harrocks, S.; Stafford, E.G.; Gardner, M.A.; Pohlner, P.G.; Tesar, P.J.; Stephens, F. The homograft aortic valve: A 29-year, 99.3% follow up of 1,022 valve replacements. J. Heart Valve Dis. 2001, 10, 334–344, discussion 5. [Google Scholar]
- Olivito, S.; Lalande, S.; Nappi, F.; Hammoudi, N.; D’Alessandro, C.; Fouret, P.; Acar, C. Structural deterioration of the cryopreserved mitral homograft valve. J. Thorac. Cardiovasc. Surg. 2012, 144, 313–320.e1. [Google Scholar] [CrossRef]
- Nappi, F.; Singh, S.S.A.; Lusini, M.; Nenna, A.; Gambardella, I.; Chello, M. The use of allogenic and autologous tissue to treat aortic valve endocarditis. Ann. Transl. Med. 2019, 7, 68. [Google Scholar] [CrossRef]
- Nappi, F.; Acar, C. Monobloc or Separate Aortic and Mitral Homografts for Endocarditis of the Intervalvular Fibrosa? Ann. Thorac. Surg. 2021, 112, 1382–1383. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Moon, M.R. A management framework for left sided endocarditis: A narrative review. Ann. Transl. Med. 2020, 8, 1627. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, U.; Spadaccio, C.; Gentile, F.; Moon, M.R.; Nappi, F. A narrative review of early surgery versus conventional treatment for infective endocarditis: Do we have an answer? Ann. Transl. Med. 2020, 8, 1626. [Google Scholar] [CrossRef] [PubMed]
- Pollari, F.; Spadaccio, C.; Cuomo, M.; Chello, M.; Nenna, A.; Fischlein, T.; Nappi, F. Sharing of decision-making for infective endocarditis surgery: A narrative review of clinical and ethical implications. Ann. Transl. Med. 2020, 8, 1624. [Google Scholar] [CrossRef] [PubMed]
- Steffen, V.; Marsch, G.; Burgwitz, K.; Kuehn, C.; Teebken, O.E. Resistance to infection of long-term cryopreserved human aortic valve allografts. J. Thorac. Cardiovasc. Surg. 2016, 151, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Heying, R.; van de Gevel, J.; Que, Y.A.; Moreillon, P.; Beekhuizen, H. Fibronectin-binding proteins and clumping factor A in Staphylococcus aureus experimental endocarditis: FnBPA is sufficient to activate human endothelial cells. Thromb. Haemost. 2007, 97, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Piroth, L.; Que, Y.A.; Widmer, E.; Panchaud, A.; Piu, S.; Entenza, J.M.; Moreillon, P. The fibrinogen- and fibronectin-binding domains of Staphylococcus aureus fibronectin-binding protein A synergistically promote endothelial invasion and experimental endocarditis. Infect. Immun. 2008, 76, 3824–3831. [Google Scholar] [CrossRef] [PubMed]
- Claes, J.; Vanassche, T.; Peetermans, M.; Liesenborghs, L.; Vandenbriele, C.; Vanhoorelbeke, K.; Missiakas, D.; Schneewind, O.; Hoylaerts, M.F.; Heying, R.; et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood 2014, 124, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Pappelbaum, K.I.; Gorzelanny, C.; Grässle, S.; Suckau, J.; Laschke, M.W.; Bischoff, M.; Bauer, C.; Schorpp-Kistner, M.; Weidenmaier, C.; Schneppenheim, R.; et al. Ultralarge von Willebrand factor fibers mediate luminal Staphylococcus aureus adhesion to an intact endothelial cell layer under shear stress. Circulation 2013, 128, 50–59. [Google Scholar] [CrossRef]
- Claes, J.; Liesenborghs, L.; Peetermans, M.; Veloso, T.R.; Missiakas, D.; Schneewind, O.; Mancini, S.; Entenza, J.M.; Hoylaerts, M.F.; Heying, R.; et al. Clumping factor A, von Willebrand factor-binding protein and von Willebrand factor anchor Staphylococcus aureus to the vessel wall. J. Thromb. Haemost. 2017, 15, 1009–1019. [Google Scholar] [CrossRef]
- Claes, J.; Ditkowski, B.; Liesenborghs, L.; Veloso, T.R.; Entenza, J.M.; Moreillon, P.; Vanassche, T.; Verhamme, P.; Hoylaerts, M.F.; Heying, R. Assessment of the Dual Role of Clumping Factor A in S. Aureus Adhesion to Endothelium in Absence and Presence of Plasma. Thromb. Haemost. 2018, 118, 1230–1241. [Google Scholar] [CrossRef]
- Ko, Y.P.; Kang, M.; Ganesh, V.K.; Ravirajan, D.; Li, B.; Höök, M. Coagulase and Efb of Staphylococcus aureus Have a Common Fibrinogen Binding Motif. mBio 2016, 7, e01885-15. [Google Scholar] [CrossRef]
- Foster, T.J. The remarkably multifunctional fibronectin binding proteins of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect Dis. 2016, 35, 1923–1931. [Google Scholar] [CrossRef]
- Ahmed, S.; Meghji, S.; Williams, R.J.; Henderson, B.; Brock, J.H.; Nair, S.P. Staphylococcus aureus fibronectin binding proteins are essential for internalization by osteoblasts but do not account for differences in intracellular levels of bacteria. Infect. Immun. 2001, 69, 2872–2877. [Google Scholar] [CrossRef] [PubMed]
- Massey, R.C.; Kantzanou, M.N.; Fowler, T.; Day, N.P.; Schofield, K.; Wann, E.R.; Berendt, A.R.; Höök, M.; Peacock, S.J. Fibronectin-binding protein A of Staphylococcus aureus has multiple, substituting, binding regions that mediate adherence to fibronectin and invasion of endothelial cells. Cell. Microbiol. 2001, 3, 839–851. [Google Scholar] [CrossRef]
- Ridley, R.A.; Douglas, I.; Whawell, S.A. Differential adhesion and invasion by Staphylococcus aureus of epithelial cells derived from different anatomical sites. J. Med. Microbiol. 2012, 61 Pt 12, 1654–1661. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niemann, S.; Nguyen, M.T.; Eble, J.A.; Chasan, A.I.; Mrakovcic, M.; Böttcher, R.T.; Preissner, K.T.; Roßlenbroich, S.; Peters, G.; Herrmann, M. More Is Not Always Better-the Double-Headed Role of Fibronectin in Staphylococcus aureus Host Cell Invasion. mBio 2021, 12, e0106221. [Google Scholar] [CrossRef]
- Pietrocola, G.; Pellegrini, A.; Alfeo, M.J.; Marchese, L.; Foster, T.J.; Speziale, P. The iron-regulated surface determinant B (IsdB) protein from Staphylococcus aureus acts as a receptor for the host protein vitronectin. J. Biol. Chem. 2020, 295, 10008–10022. [Google Scholar] [CrossRef]
- Alfeo, M.J.; Pagotto, A.; Barbieri, G.; Foster, T.J.; Vanhoorelbeke, K.; De Filippis, V.; Speziale, P.; Pietrocola, G. Staphylococcus aureus iron-regulated surface determinant B (IsdB) protein interacts with von Willebrand factor and promotes adherence to endothelial cells. Sci. Rep. 2021, 11, 22799. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, K.; Ishikawa, M.; Morita, Y.; Yokogawa, N.; Xie, C.; de Mesy Bentley, K.L.; Ito, H.; Kates, S.L.; Daiss, J.L.; Schwarz, E.M. IsdB antibody-mediated sepsis following S. aureus surgical site infection. JCI Insight 2020, 5, e141164. [Google Scholar] [CrossRef]
- Tsai, C.M.; Caldera, J.R.; Hajam, I.A.; Chiang, A.W.T.; Tsai, C.H.; Li, H.; Díez, M.L.; Gonzalez, C.; Trieu, D.; Martins, G.A.; et al. Non-protective immune imprint underlies failure of Staphylococcus aureus IsdB vaccine. Cell Host Microbe. 2022, 30, 1163–1172.e6. [Google Scholar] [CrossRef]
- Leeten, K.; Jacques, N.; Lancellotti, P.; Oury, C. Aspirin or Ticagrelor in Staphylococcus aureus Infective Endocarditis: Where Do We Stand? Front. Cell Dev. Biol. 2021, 9, 716302. [Google Scholar] [CrossRef]
- Ditkowski, B.; Bezulska-Ditkowska, M.; Jashari, R.; Baatsen, P.; Moreillon, P.; Rega, F.; Veloso, T.R.; Hoylaerts, M.F.; Heying, R.; Congenital Cardiology and Cardiac Surgery Group. Antiplatelet therapy abrogates platelet-assisted Staphylococcus aureus infectivity of biological heart valve conduits. Congenital Cardiology and Cardiac Surgery Group. J. Thorac. Cardiovasc. Surg. 2021, 161, e457–e472. [Google Scholar] [CrossRef] [PubMed]
- Hannachi, N.; Habib, G.; Camoin-Jau, L. Aspirin Effect on Staphylococcus aureus Platelet Interactions During Infectious Endocarditis. Front. Med. 2019, 6, 217. [Google Scholar] [CrossRef] [PubMed]
- Park ENa, H.S.; Song, Y.R.; Shin, S.Y.; Kim, Y.M.; Chung, J. Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect. Immun. 2014, 82, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Veloso, T.R.; Que, Y.A.; Chaouch, A.; Giddey, M.; Vouillamoz, J.; Rousson, V.; Moreillon, P.; Entenza, J.M. Prophylaxis of experimental endocarditis with antiplatelet and antithrombin agents: A role for long-term prevention of infective endocarditis in humans? J. Infect. Dis. 2015, 11, 72–79. [Google Scholar] [CrossRef]
- Braï, M.A.; Hannachi, N.; El Gueddari, N.; Baudoin, J.P.; Dahmani, A.; Lepidi, H.; Habib, G.; Camoin-Jau, L. The Role of Platelets in Infective Endocarditis. Int. J. Mol. Sci. 2023, 24, 7540. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Avtaar Singh, S.S. Host–Bacterium Interaction Mechanisms in Staphylococcus aureus Endocarditis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 11068. https://doi.org/10.3390/ijms241311068
Nappi F, Avtaar Singh SS. Host–Bacterium Interaction Mechanisms in Staphylococcus aureus Endocarditis: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(13):11068. https://doi.org/10.3390/ijms241311068
Chicago/Turabian StyleNappi, Francesco, and Sanjeet Singh Avtaar Singh. 2023. "Host–Bacterium Interaction Mechanisms in Staphylococcus aureus Endocarditis: A Systematic Review" International Journal of Molecular Sciences 24, no. 13: 11068. https://doi.org/10.3390/ijms241311068
APA StyleNappi, F., & Avtaar Singh, S. S. (2023). Host–Bacterium Interaction Mechanisms in Staphylococcus aureus Endocarditis: A Systematic Review. International Journal of Molecular Sciences, 24(13), 11068. https://doi.org/10.3390/ijms241311068