


Metagenomic Insights Reveal the Microbial Diversity and Associated Algal-Polysaccharide-Degrading Enzymes on the Surface of Red Algae among Remote Regions
Abstract
1. Introduction
2. Results and Discussion
2.1. Characteristics of the Nine Macroalgae Samples
2.2. Sequencing and Metagenomic Assembly
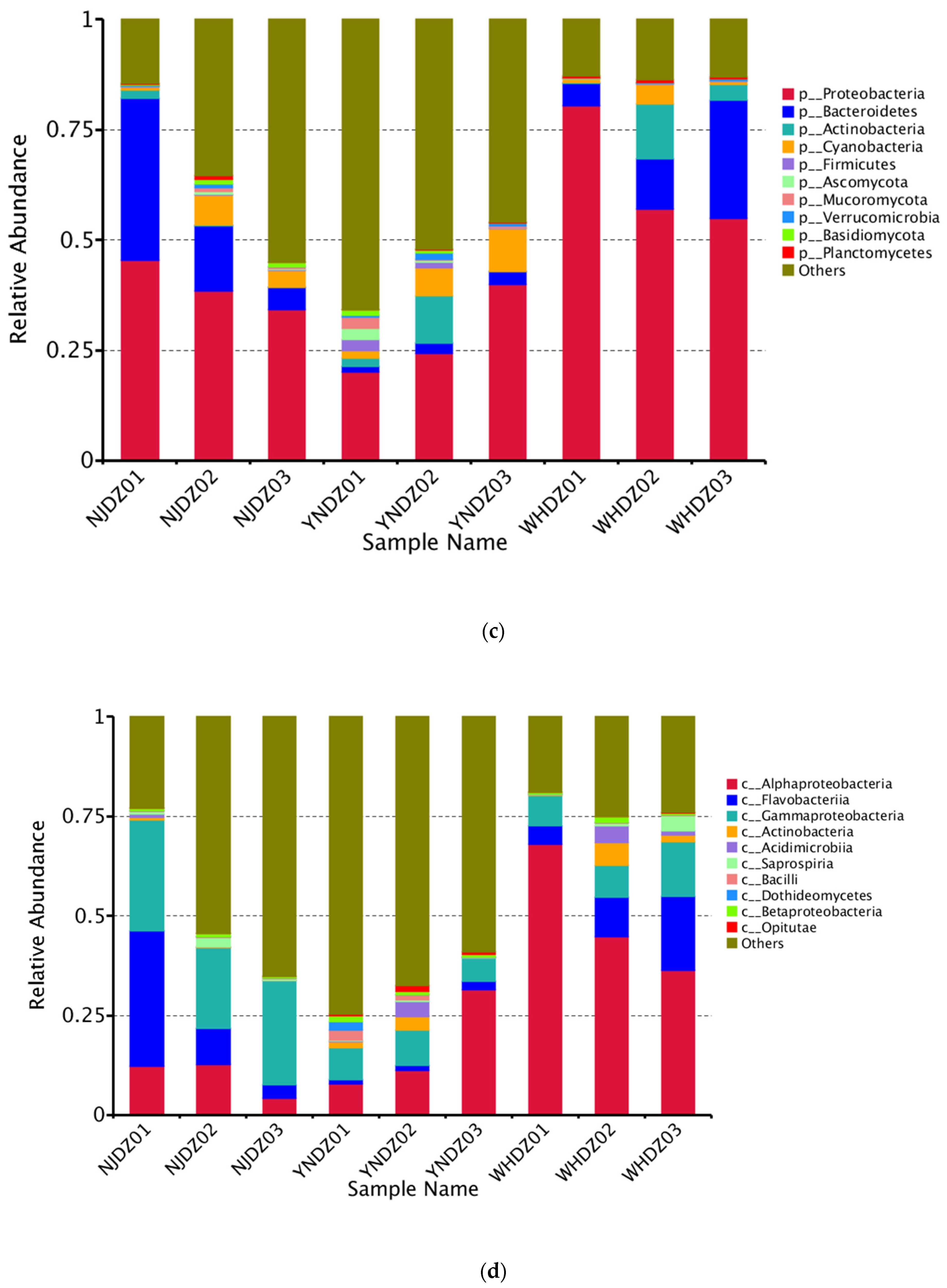
2.3. Microbial Diversity and Community Composition
2.4. Functional Prediction of Bacterial Communities
2.5. CAZymes Insights on the Bacterial Community
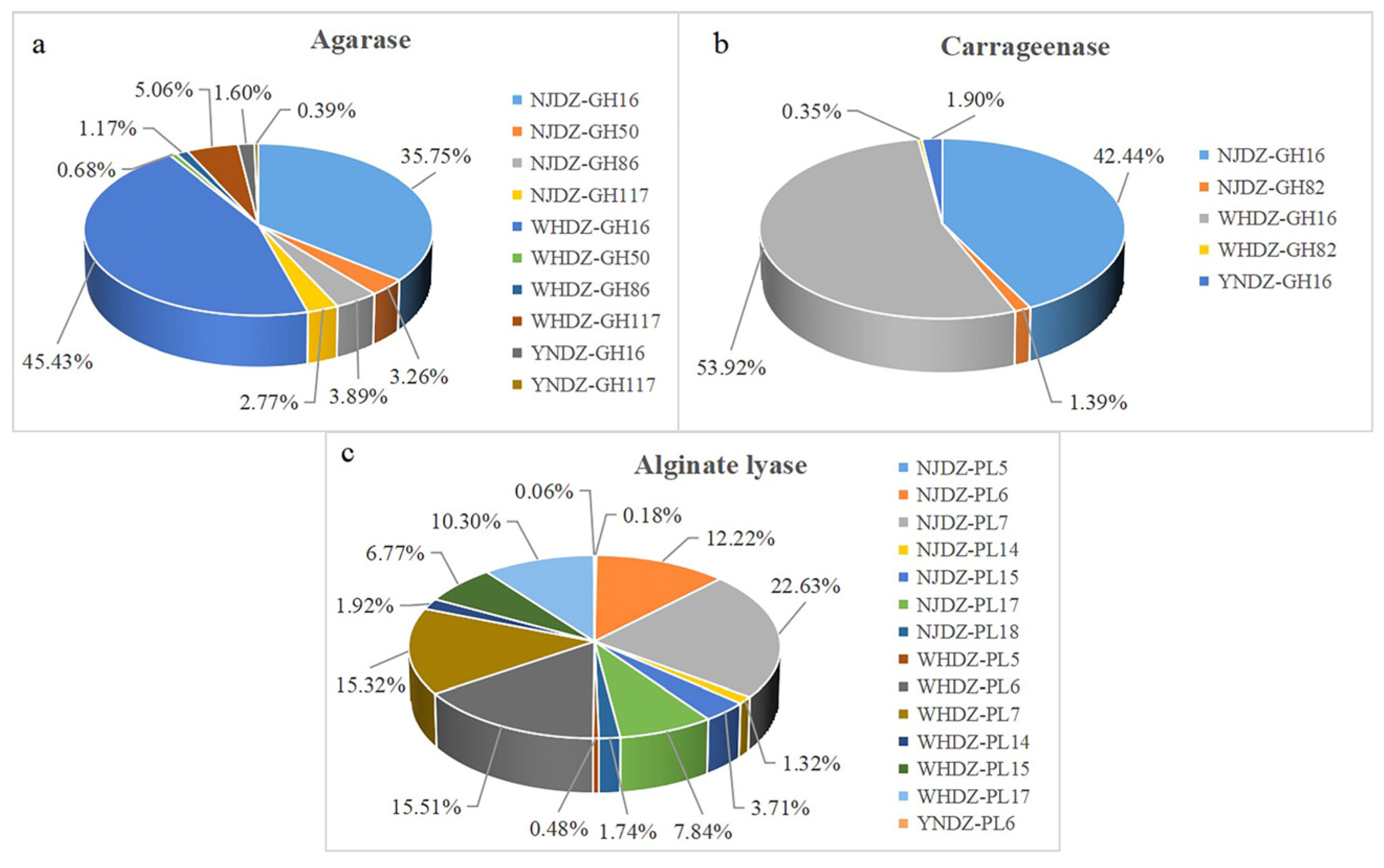
2.6. Mining of Enzymes for Algal Polysaccharide Degradation
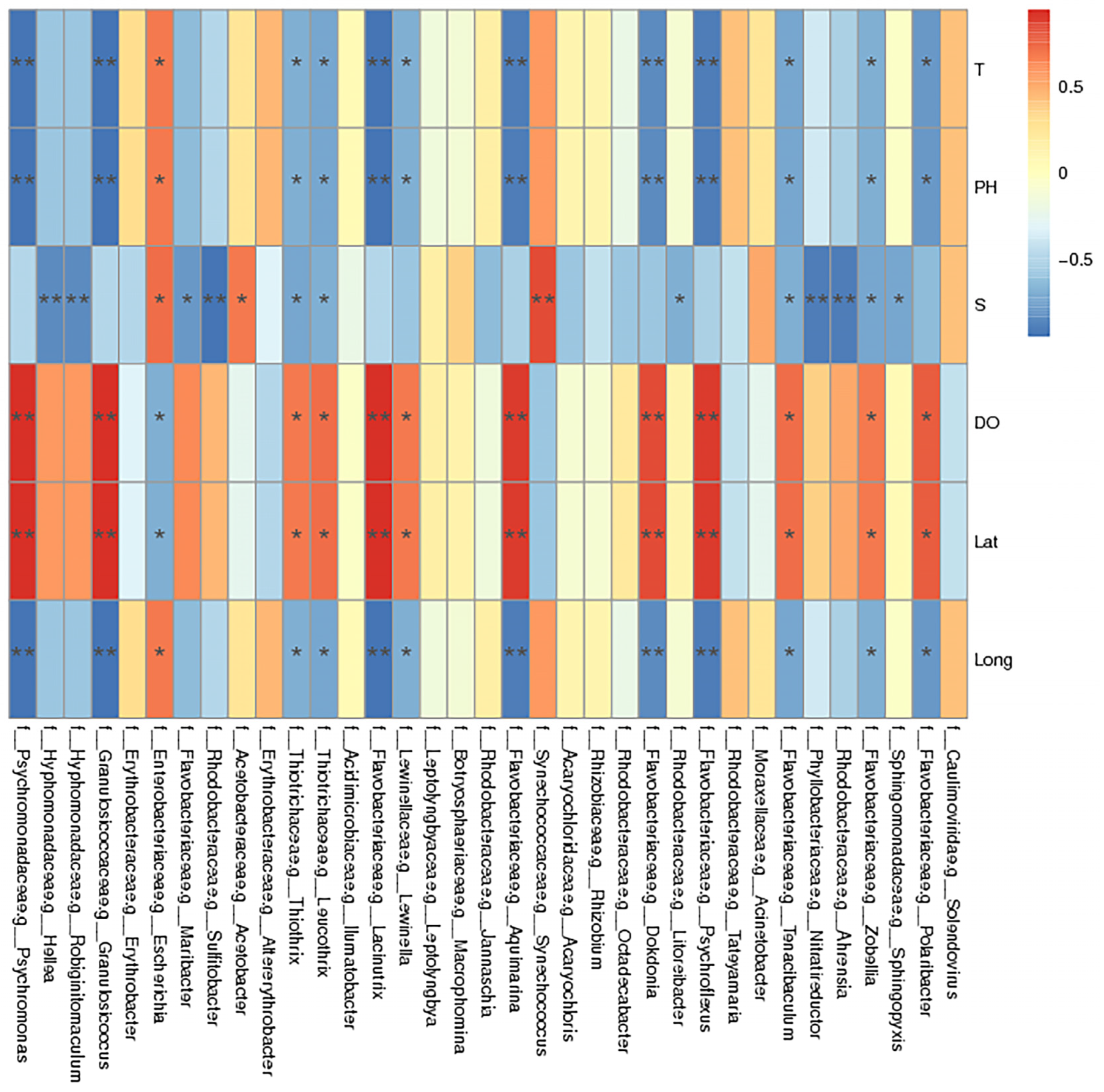
2.7. Environmental Factor Analysis
2.8. Diverse Bacterial Lineages Potentially Harbor Antibiotic Resistance Genes
3. Materials and Methods
3.1. Macroalgae Sampling and Characterization
3.2. DNA Extraction and Sequencing
3.3. Metagenome Assembly
3.4. Gene Prediction and Abundance Analysis
3.5. Taxonomy Prediction
3.6. Functional Annotations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Field, C.B.; Behrenfeld, M.J.; Randerson, J.T.; Falkowski, P. Primary production of the biosphere: Integrating terrestrial and oceanic components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef]
- Hedges, J.I.; Baldock, J.A.; Ge’linas, Y.; Lee, C.; Peterson, M.; Wakeham, S.G. Evidence for non-selective preservation of organic matter in sinking marine particles. Nature 2001, 409, 801–804. [Google Scholar] [CrossRef]
- Martin, M.; Portetelle, D.; Michel, G.; Vandenbol, M. Microorganisms living on macroalgae: Diversity, interactions, and biotechnological applications. Appl. Microbiol. Biot. 2014, 98, 2917–2935. [Google Scholar] [CrossRef]
- Cundell, A.M.; Sleeter, T.D.; Mitchell, R. Microbial populations associated with the surface of the brown alga Ascophyllum nodosum. Microb. Ecol. 1997, 4, 81–91. [Google Scholar] [CrossRef]
- Wietz, M.; Wemheuer, B.; Simon, H.; Giebel, H.-A.; Seibt, M.A.; Daniel, R.; Brinkhoff, T.; Simon, M. Bacterial community dynamics during polysaccharide degradation at contrasting sites in the Southern and Atlantic Oceans. Environ. Microbiol. 2015, 17, 3822–3831. [Google Scholar] [CrossRef]
- Gu, X.Q.; Zhao, L.Y.; Tan, J.J.; Zhang, Q.; Fu, L.P.; Li, J. Characterization of a novel β-agarase from Antarctic macroalgae-associated bacteria metagenomic library and anti-inflammatory activity of the enzymatic hydrolysates. Front. Microbiol. 2022, 13, 972272. [Google Scholar] [CrossRef]
- Hanson, A.D.; Pribat, A.; Waller, J.C.; Cre’cy-Lagard, V. “Unknown” proteins and “orphan” enzymes: The missing half of the engineering parts list—And how to find it. Biochem. J. 2010, 425, 1–11. [Google Scholar] [CrossRef]
- Popper, Z.A.; Michel, G.; Hervé, C.; Domozych, D.S.; Willats, W.G.T.; Tuohy, M.G.; Kloareg, B.; Stengel, D.B. Evolution and diversity of plant cell walls: From algae to flowering plants. Annu. Rev. Plant. Biol. 2011, 62, 567–590. [Google Scholar] [CrossRef]
- Chiovitti, A.; Kraft, G.T.; Bacic, A.; Liao, M.L. Review: Gelling polysaccharides from Australian seaweeds: Research and potential. Mar. Freshw. Res. 2001, 52, 917–935. [Google Scholar] [CrossRef]
- Sarwar, G.; Matayoshi, S.; Oda, H. Purification of a kappacarrageenase from marine Cytophaga species. Microbiol. Immunol. 2013, 31, 869–877. [Google Scholar] [CrossRef]
- Blackburn, N.; Fenchel, T.; Mitchell, J. Microscale nutrient patches in planktonic habitats shown by chemotactic bacteria. Science 1998, 282, 254–2256. [Google Scholar] [CrossRef]
- Arnosti, C.; Wietz, M.; Brinkhoff, T.; Hehemann, J.-H.; Probandt, D.; Zeugner, L.; Amann, R. The biogeochemistry of marine polysaccharides: Sources, inventories, and bacterial drivers of the carbohydrate cycle. Annu. Rev. Mar. Sci. 2021, 13, 81–108. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Kennaway, G.; Franca, S.; Alverca, E. Bacterium-dinoflagellate interactions: Investigative microscopy of Alexandrium spp. (Gonyaulacales, Dinophyceae). Phycologia 2001, 40, 280–285. [Google Scholar] [CrossRef]
- Bell, W.; Mitchell, R. Chemotactic and growth responses of marine bacteria to algal extracellular products. Biol. Bull. 1972, 143, 265–277. [Google Scholar] [CrossRef]
- Ohta, S.; Chang, T.; Ikegami, N.; Kondo, M.; Miyata, H. Antibiotic substance produced by a newly isolated marine microalga, Chlorococcum HS-101. B. Arch. Environ. Contam. Toxicol. 1993, 50, 171–178. [Google Scholar] [CrossRef]
- Kubanek, J.; Jensen, P.R.; Keifer, P.A.; Sullards, M.C.; Collins, D.O.; Fenical, W. Seaweed resistance to microbial attack: A targeted chemical defense against marine fungi. Proc. Natl. Acad. Sci. USA 2003, 100, 6916–6921. [Google Scholar] [CrossRef]
- Lachnit, T.; Blümel, M.; Imhoff, J.; Wahl, M. Specific epibacterial communities on macroalgae: Phylogeny matters more than habitat. Aquat. Biol. 2009, 5, 181–186. [Google Scholar] [CrossRef]
- A Tujula, N.; Crocetti, G.R.; Burke, C.; Thomas, T.; Holmström, C.; Kjelleberg, S. Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J. 2010, 4, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Hengst, M.B.; Andrade, S.; González, B.; Correa, J.A. Changes in epiphytic bacterial communities of intertidal seaweeds modulated by host, temporality, and copper enrichment. Microb. Ecol. 2010, 60, 282–290. [Google Scholar] [CrossRef]
- Campbell, A.H.; Marzinelli, E.M.; Gelber, J.; Steinberg, P.D. Spatial variability of microbial assemblages associated with a dominant habitatforming seaweed. Front. Microbiol. 2015, 6, 230. [Google Scholar] [CrossRef]
- Joke, H.; Frederik, L.; Olivier, D.C.; Anne, W. What we can learn from sushi: A review on seaweed-bacterial associations. Fems. Microbiol. Ecol. 2012, 83, 1–16. [Google Scholar]
- Gui, Y.Y.; Gu, X.Q.; Fu, L.P.; Zhang, Q.; Fu, L.P.; Li, J. Expression and characterization of a thermostable carrageenase from an Antarctic Polaribacter sp. NJDZ03 strain. Front. Microbiol. 2021, 12, 631039. [Google Scholar] [CrossRef]
- Li, J.; Pan, A.H.; Xie, M.S.; Zhang, P.P.; Gu, X.Q. Characterization of a thermostable κ-carrageenase from a hot spring bacterium and plant protection activity of the oligosaccharide enzymolysis product. J. Sci. Food Agric. 2019, 99, 1812–1819. [Google Scholar] [CrossRef]
- Hehemann, J.H.; Boraston, A.B.; Czjzek, M. A sweet new wave: Structures and mechanisms of enzymes that digest polysaccharides from marine algae. Curr. Opin. Struc. Biol. 2014, 28, 7–86. [Google Scholar] [CrossRef]
- Bhattacharyya, P.N.; Tanti, B.; Barman, P.; Jhn, D.K. Culture-independent metagenomic approach to characterize the surface and subsurface soil bacterial community in the Brahmaputra valley, Assam, North-East India, an Indo-Burma mega-biodiversity hotspot. World J. Microb. Biot. 2014, 30, 519–528. [Google Scholar] [CrossRef]
- Tian, X.P.; Long, L.J.; Wang, F.Z.; Xu, Y.; Li, J.; Zhang, J.; Zhang, C.S.; Zhang, S.; Li, W.J. Streptomyces nanhaiensis sp. nov. a marine streptomycete isolated from a deep-sea sediment. Int. J. Syst. Evol. Microbiol. 2012, 62, 864–868. [Google Scholar] [CrossRef]
- Tringe, S.G.; Rubin, E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 2005, 6, 805–814. [Google Scholar] [CrossRef]
- Pasmore, M.; Costerton, J.W. Biofilms, bacterial signaling, and their ties to marine biology. J. Ind. Microbiol. Biotechnol. 2003, 30, 407–413. [Google Scholar] [CrossRef]
- Cottrell, M.T.; Kirchman, D.L. Natural assemblages of marine proteobacteria and members of the cytophaga-flavobacter cluster consuming low-and high-molecular-weight dissolved organic matter. Appl. Environ. Microbiol. 2000, 66, 1692–1697. [Google Scholar] [CrossRef]
- Segawa, T.; Miyamoto, K.; Ushida, K.; Agata, K.; Okada, N.; Kohshima, S. Seasonal change in bacterial flora and biomass in mountain snow from the Tateyama Mountains, Japan, analyzed by 16S rRNA gene sequencing and real-time PCR. Appl. Environ. Microbiol. 2005, 71, 123–130. [Google Scholar] [CrossRef]
- Amato, P.; Hennebelle, R.; Magand, O.; Sancelme, M.; Delort, A.M.; Barbante, C. Bacterial characterization of the snow cover at Spitzberg, Svalbard. FEMS Microbiol. Ecol. 2007, 59, 255–264. [Google Scholar] [CrossRef]
- Viera, R.P.; Ggonzalez, A.M.; Cardoso, A.M. Relationships between bacterial diversity and environmental variables in a tropical marine environment, Rio de Janeiro. Environ. Microbiol. 2008, 10, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Lozada, M.; Diéguez, M.C.; García, P.E.; Dionisi, H.M. Microbial communities associated with kelp detritus in temperate and subantarctic intertidal sediments. Sci. Total Environ. 2023, 857, 159392. [Google Scholar] [CrossRef]
- Martin, K.; Schmidt, K.; Toseland, A.; Boulton, C.A.; Barry, K.; Beszteri, B.; Brussaard, C.P.D.; Clum, A.; Daum, C.G.; Eloe-Fadrosh, E.; et al. The biogeographic differentiation of algal microbiomes in the upper ocean from pole to pole. Nat. Commun. 2021, 12, 5483. [Google Scholar] [CrossRef]
- Martin, M.; Barbeyron, T.; Martin, R.; Portetelle, D.; Michel, G.; Vandenbol, M. The cultivable surface microbiota of the brown alga Ascophyllum nodosum is enriched in macroalgal-polysaccharide-degrading bacteria. Front. Microbiol. 2015, 6, 1487. [Google Scholar] [CrossRef]
- Naretto, A.; Fanuel, M.; Ropartz, D.; Rogniaux, H.; Larocque, R.; Czjzek, M.; Tellier, C.; Michel, G. The agar-specific hydrolase Zg AgaC from the marine bacterium Zobellia galactanivorans defines a new GH16 protein subfamily. J. Biol. Chem. 2019, 294, 6923–6939. [Google Scholar] [CrossRef]
- Egelund, J.; Ellis, M.; Doblin, M.; Qu, Y.; Bacic, A. Genes and enzymes of the GT31 family: Towards unravelling the function(s) of the plant glycosyltransferase family members. In Annual Plant Reviews: Plant Polysaccharides, Biosynthesis and Bioengineering; Wiley-Blackwell: Hoboken, NJ, USA, 2010. [Google Scholar]
- Matard-Mann, M.; Bernard, T.; Leroux, C.; Barbeyron, T.; Larocque, R.; Préchoux, A.; Jeudy, A.; Jam, M.; Collén, P.N.; Michel, G.; et al. Structural insights into marine carbohydrate degradation by family GH16 κ-carrageenases. J. Biol. Chem. 2017, 292, 19919–19934. [Google Scholar] [CrossRef]
- Pomin, V.H. Structural and functional insights into sulfated galactans: A systematic review. Glycoconj. J. 2009, 27, 1–12. [Google Scholar] [CrossRef]
- Lavin, P.; Gallardo-Cerda, J.; Torres-Diaz, C.; Asencio, G.; Gonzalez, M. Antarctic strain of Bacillus sp. with extracellular agarolitic and alginate-lyase activities. Gayana 2013, 77, 75–82. [Google Scholar]
- Dhargalkar, V.K.; Verlecar, X.N. Southern Ocean seaweeds: A resource for exploration in food and drugs. Aquaculture 2009, 287, 229–242. [Google Scholar] [CrossRef]
- Zhu, B.; Ni, F.; Xiong, Q.; Yao, Z. Marine oligosaccharides originated from seaweeds: Source, preparation, structure, physiological activity and applications. Crit. Rev. Food Sci. 2021, 61, 60–74. [Google Scholar] [CrossRef]
- Herrmann, M.; Saunders, A.M.; Schramm, A. Archaea dominate the ammonia-oxidizing community in the rhizosphere of the freshwater macrophyte Littorella uniflora. Appl. Environ. Microbiol. 2008, 74, 3279–3283. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Wu, H.; Cao, Y.; Xing, W.; Jeppesen, E.; Li, W. Comparison of periphyton communities on natural and artificial macrophytes with contrasting morphological structures. Freshw. Biol. 2017, 62, 1783–1793. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Ibarbalz, F.M.; Henry, N.; Brandão, M.C.; Martini, S.; Busseni, G.; Byrne, H.; Coelho, L.P.; Endo, H.; Gasol, J.M.; Gregory, A.C.; et al. Global trends in marine plankton diversity across kingdoms of life. Cell 2019, 179, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.S.; Isbell, F.; Seidl, R. Beta-diversity, community assembly, and ecosystem functioning. Trends Ecol. Evol. 2018, 33, 549–564. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Song, L.; Wu, J.; Sun, W.; Teng, Y. Efects on microbiomes and resistomes and the source-specifc ecological risks of heavy metals in the sediments of an urban river. J. Hazard. Mater. 2021, 409, 124472. [Google Scholar] [CrossRef] [PubMed]
- Imchen, M.; Kumavath, R. Metagenomic insights into the antibiotic resistome of mangrove sediments and their association to socioeconomic status. Environ. Pollut. 2021, 268, 115795. [Google Scholar] [CrossRef]
- Zhao, R.; Yu, K.; Zhang, J.; Zhang, G.; Huang, J.; Ma, L.; Deng, C.; Li, X.; Li, B. Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res. 2020, 186, 116318. [Google Scholar] [CrossRef]
- Cuadrat, R.R.C.; Sorokina, M.; Andrade, B.; Goris, T.; Dávila, A.M.R. Global ocean resistome revealed: Exploring antibiotic resistance gene abundance and distribution in TARA Oceans samples. GigaSciencem 2020, 9, giaa046. [Google Scholar] [CrossRef]
- Chen, J.; McIlroy, S.E.; Archana, A.; Baker, D.M.; Panagiotou, G. A pollution gradient contributes to the taxonomic, functional, and resistome diversity of microbial communities in marine sediments. Microbiome 2019, 7, 104. [Google Scholar] [CrossRef]
- Dong, X.Y.; Lan, H.Y.; Huang, L.G. Metagenomic views of microbial communities in sand sediments associated with coral reefs. Microb. Ecol. 2022, 85, 465–477. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Chatelier, E.L.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids. Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldón, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4. 0: Nested orthology inference across 3686 organisms. Nucleic Acids. Res. 2013, 42, D231–D239. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids. Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Lat | Long | DO | pH | Salinity | Temperature | The Dominant Macroalgae-Associated Bacteria |
|---|---|---|---|---|---|---|---|
| Great Wall Station | 62.2° | 58.95° | 10.836 mg/L | 8.01 | 34.07‰ | −1.34 °C | Gammaproteobacteria, Flavobacteriia, Alphaproteobacteria, Saprospiria |
| Halmahera | 0.6° | 127.86° | 5.44 mg/L | 8.79 | 34.7‰ | 27.9 °C | Alphaproteobacteria, Gammaproteobacteria, Actinobacteria, Flavobacteriia |
| Weihai | 37.42° | 122.28° | 6.65 mg/L | 8.43 | 32.4‰ | 8 °C | Alphaproteobacteria, Flavobacteriia, Gammaproteobacteria, Actinobacteria |
| Gene catalogue | 2,485,201 |
| Annotated on KEGG | 1,493,359 (60.09%) |
| Annotated on KO | 806,765 (32.46%) |
| Annotated on KO number | 7566 |
| Annotated on EC | 493,520 (19.86%)/2517 |
| Annotated on pathway | 507,914 (20.44%)/409 |
| Annotated on eggNOG | 1,428,379 (57.48%) |
| Annotated on OG | 1,428,379 (57.48%)/31,380 |
| Annotated on CAZy | 61,807 (2.49%) |
| Location | Enzyme Number | E.C. Number | CAZy Family |
|---|---|---|---|
| Agarase | |||
| NJDZ | 939 | EC 3.2.1.81 | GH16 |
| EC 3.2.1.81 | GH50 | ||
| EC 3.2.1.81 | GH86 | ||
| EC 3.2.1.- | GH117 | ||
| WHDZ | 1076 | EC 3.2.1.81 | GH16 |
| EC 3.2.1.81 | GH50 | ||
| EC 3.2.1.81 | GH86 | ||
| EC 3.2.1.- | GH117 | ||
| YNDZ | 41 | EC 3.2.1.81 | GH16 |
| EC 3.2.1.- | GH117 | ||
| Carrageenase | |||
| NJDZ | 759 | EC 3.2.1.83 | GH16 |
| EC 3.2.1.157 | GH82 | ||
| WHDZ | 940 | EC 3.2.1.83 | GH16 |
| EC 3.2.1.157 | GH82 | ||
| YNDZ | 33 | EC 3.2.1.83 | GH16 |
| Alginate lyase | |||
| NJDZ | 829 | EC 4.2.2.3 | PL5 |
| EC 4.2.2.3 | PL6 | ||
| EC 4.2.2.- | PL7 | ||
| EC 4.2.2.3 | PL14 | ||
| EC 4.2.2.3 | PL15 | ||
| EC 4.2.2.3 | PL17 | ||
| EC 4.2.2.3 | PL18 | ||
| WHDZ | 840 | EC 4.2.2.3 | PL5 |
| EC 4.2.2.3 | PL6 | ||
| EC 4.2.2.- | PL7 | ||
| EC 4.2.2.3 | PL14 | ||
| EC 4.2.2.3 | PL15 | ||
| EC 4.2.2.3 | PL17 | ||
| YNDZ | 1 | EC 4.2.2.3 | PL6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, X.; Cao, Z.; Zhao, L.; Seswita-Zilda, D.; Zhang, Q.; Fu, L.; Li, J. Metagenomic Insights Reveal the Microbial Diversity and Associated Algal-Polysaccharide-Degrading Enzymes on the Surface of Red Algae among Remote Regions. Int. J. Mol. Sci. 2023, 24, 11019. https://doi.org/10.3390/ijms241311019
Gu X, Cao Z, Zhao L, Seswita-Zilda D, Zhang Q, Fu L, Li J. Metagenomic Insights Reveal the Microbial Diversity and Associated Algal-Polysaccharide-Degrading Enzymes on the Surface of Red Algae among Remote Regions. International Journal of Molecular Sciences. 2023; 24(13):11019. https://doi.org/10.3390/ijms241311019
Chicago/Turabian StyleGu, Xiaoqian, Zhe Cao, Luying Zhao, Dewi Seswita-Zilda, Qian Zhang, Liping Fu, and Jiang Li. 2023. "Metagenomic Insights Reveal the Microbial Diversity and Associated Algal-Polysaccharide-Degrading Enzymes on the Surface of Red Algae among Remote Regions" International Journal of Molecular Sciences 24, no. 13: 11019. https://doi.org/10.3390/ijms241311019
APA StyleGu, X., Cao, Z., Zhao, L., Seswita-Zilda, D., Zhang, Q., Fu, L., & Li, J. (2023). Metagenomic Insights Reveal the Microbial Diversity and Associated Algal-Polysaccharide-Degrading Enzymes on the Surface of Red Algae among Remote Regions. International Journal of Molecular Sciences, 24(13), 11019. https://doi.org/10.3390/ijms241311019