
Tumor Site-Specific Cleavage Improves the Antitumor Efficacy of Antibody–Drug Conjugates


Abstract

1. Introduction
2. Results and Discussion
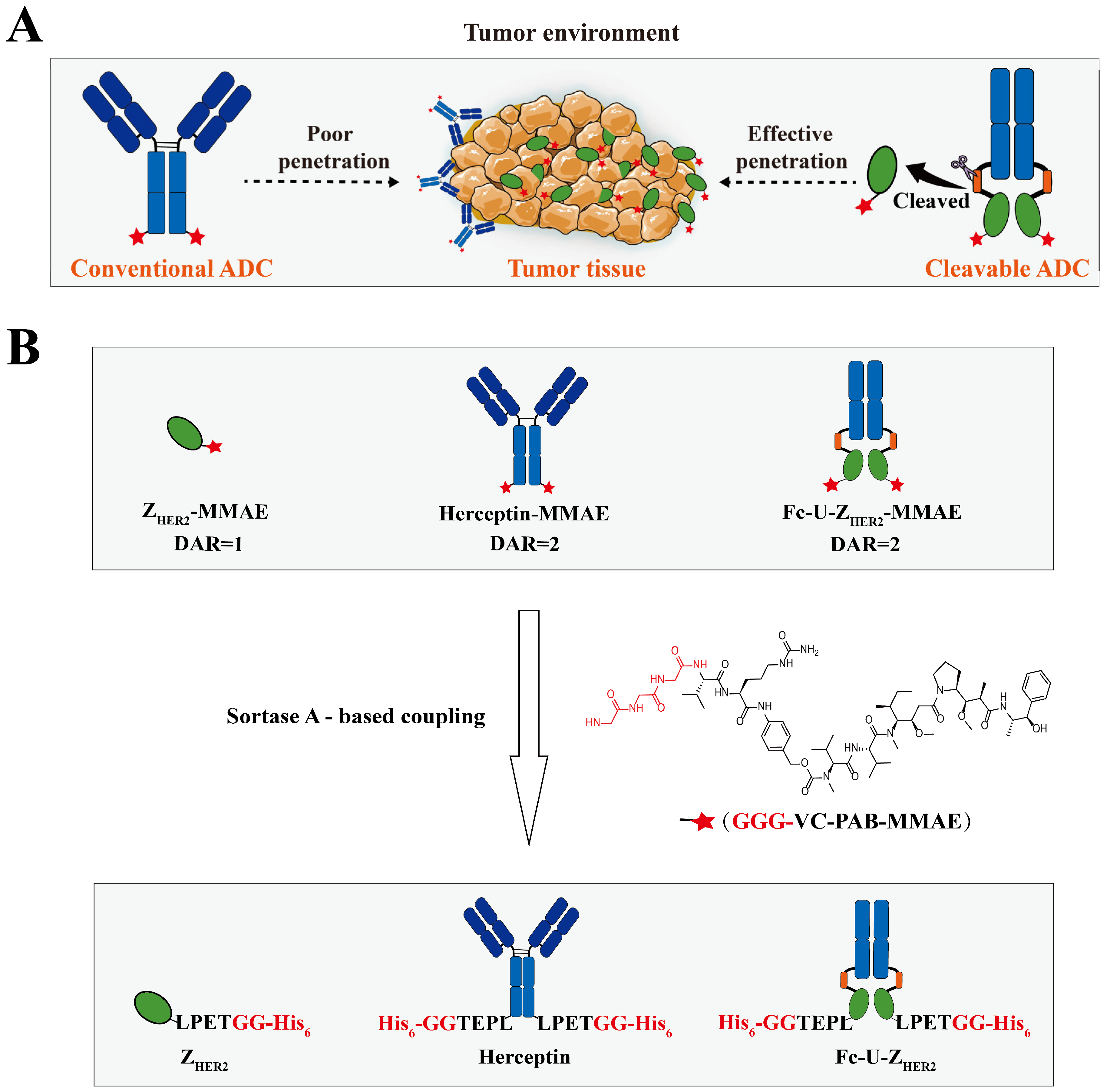
2.1. Design and Expression of the Raw Proteins
2.2. Enzymatic Coupling of MMAE Catalyzed by Sortase A
2.3. Ability of Fc-U-ZHER2-MMAE to Bind to the HER2 Receptor
2.4. Subcellular Colocalization of the Conjugates Determined by FACS and Confocal Microscopy
2.5. Cytotoxicity Analysis of Fc-U-ZHER2-MMAE Determined by MTT Assays and FACS
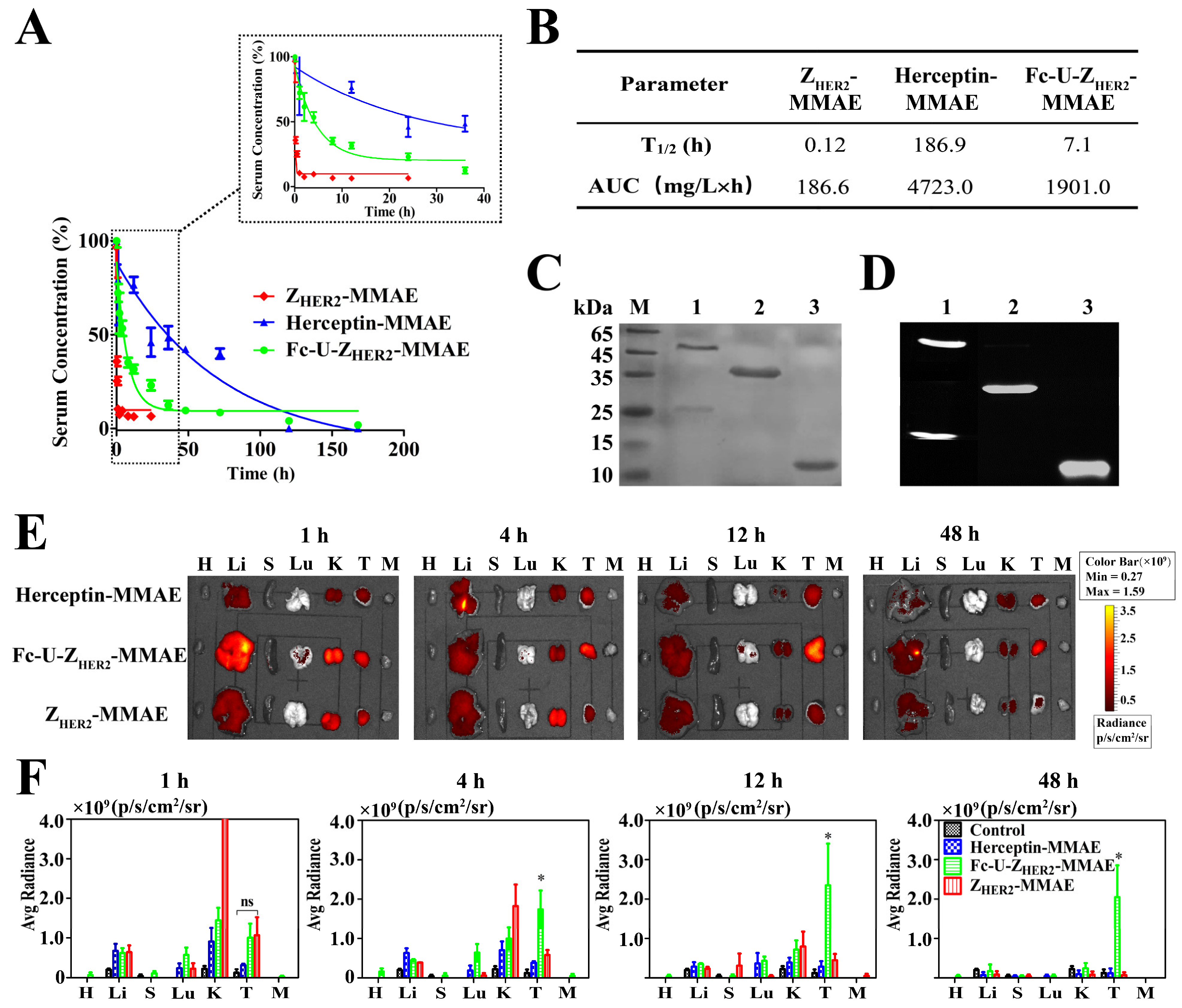
2.6. Pharmacokinetics and Biodistribution of the Conjugates in Mice
2.7. Detection of uPA Enzyme Distribution and Activity
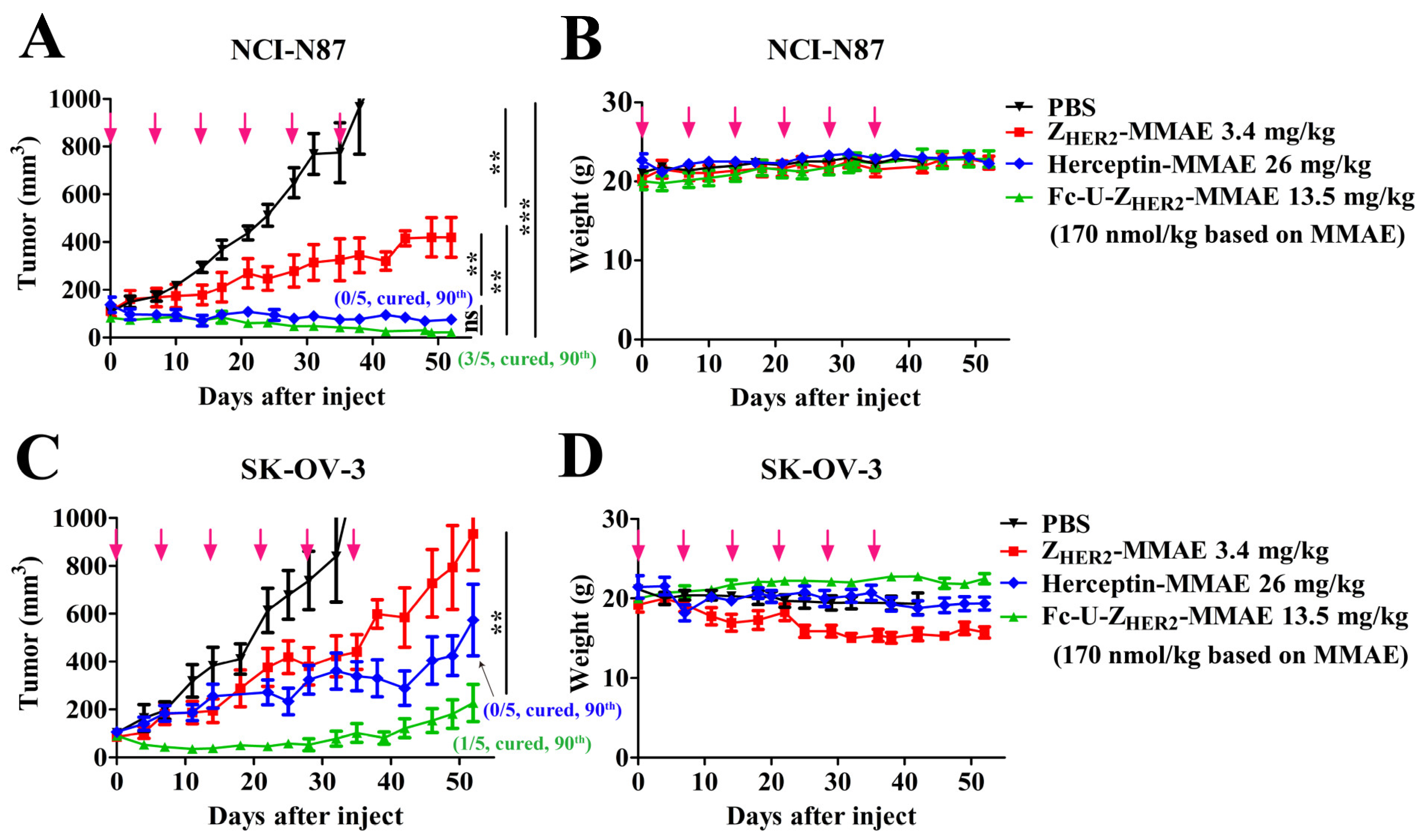
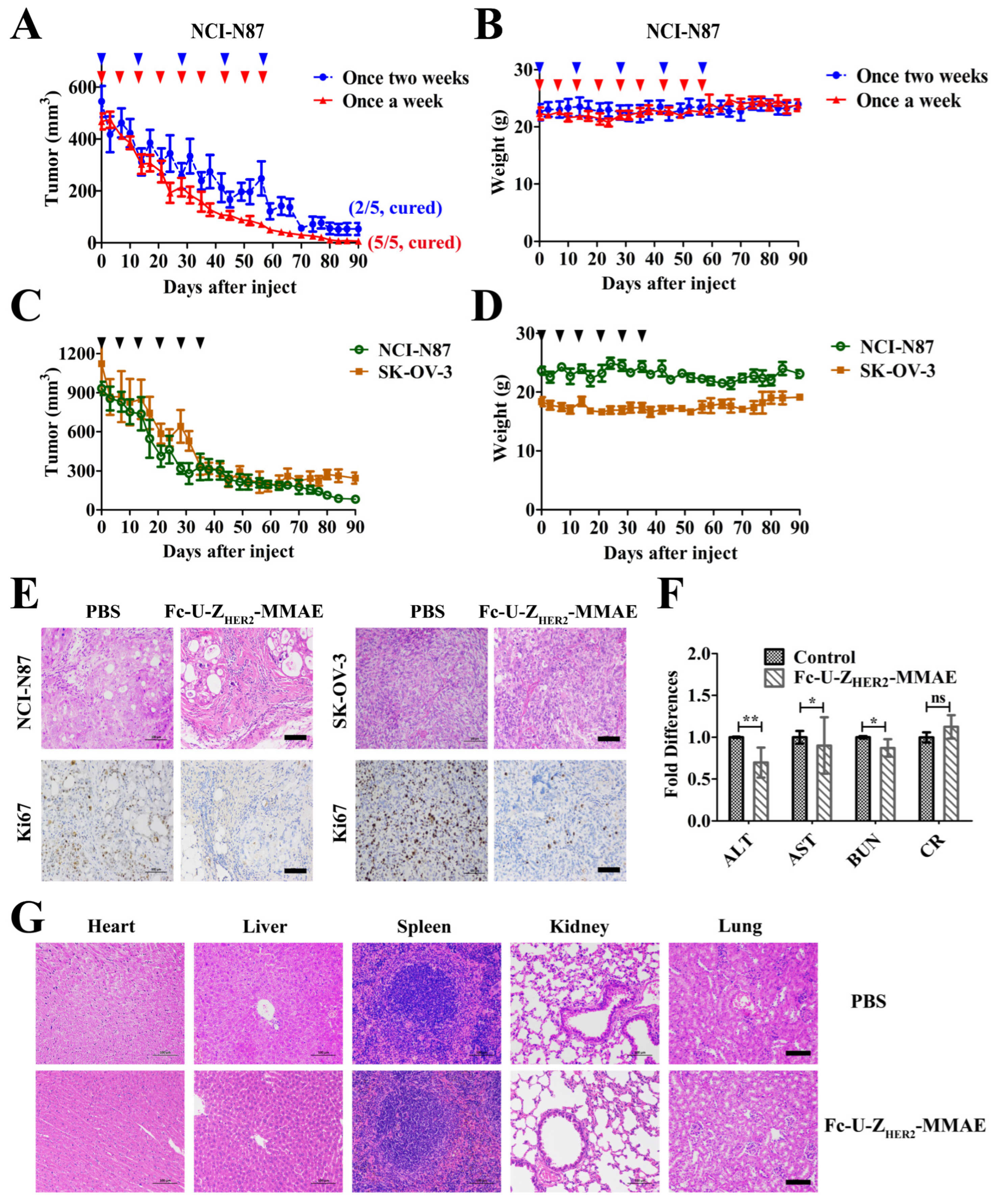
2.8. In Vivo Antitumor Efficacy of the Conjugates
2.9. Off-Target Toxicity Analysis
3. Materials and Methods
3.1. Reagents
3.2. Cell Lines and Animals
3.3. Expression and Purification of the Proteins with LPETGG- 6 × His Tag
3.4. Preparation and Purification of the Conjugates Based on Sortase A
3.5. Analysis of Binding Affinity Based on ELISA
3.6. In Vitro Cytotoxicity Assays
3.7. In Vivo Pharmacokinetics and Biodistribution
3.8. In Vivo Antitumor Activity
3.9. Detection of Biochemical Indices and H&E and Ki-67 Staining Post-Treatment
3.10. In Vivo Off-Target Toxicity Analysis
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-drug conjugates: A comprehensive review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Yu, J.; Fang, T.; Yun, C.; Liu, X.; Cai, X. Antibody-drug conjugates targeting the human epidermal growth factor receptor family in cancers. Front. Mol. Biosci. 2022, 9, 847835. [Google Scholar] [CrossRef]
- Epenetos, A.A.; Snook, D.; Durbin, H.; Johnson, P.M.; Taylor-Papadimitriou, J. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res. 1986, 46, 3183–3191. [Google Scholar]
- Wang, X.; Zhu, J.; Zhao, P.; Jiao, Y.; Xu, N.; Grabinski, T.; Liu, C.; Miranti, C.K.; Fu, T.; Cao, B. In vitro efficacy of immuno-chemotherapy with anti-EGFR human fab-taxol conjugate on A431 epidermoid carcinoma cells. Cancer Biol. Ther. 2007, 6, 980–987. [Google Scholar] [CrossRef]
- Chen, X.; Ding, G.; Gao, Q.; Sun, J.; Zhang, Q.; Du, L.; Qiu, Z.; Wang, C.; Zheng, F.; Sun, B.; et al. A human anti-c-Met Fab fragment conjugated with doxorubicin as targeted chemotherapy for hepatocellular carcinoma. PLoS ONE 2013, 8, e63093. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.-H.; Li, Y.-H.; Jiang, Y.; Xie, P.-L.; Zhou, G.-H.; Li, G.-C. Anti-hepatoma human single-chain Fv antibody and adriamycin conjugates with potent antitumor activity. Int. Immunopharmacol. 2014, 18, 20–26. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Designed ankyrin repeat proteins (darpins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Simon, M.; Frey, R.; Zangemeister-Wittke, U.; Plückthun, A. Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjugate Chem. 2013, 24, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Ghanemi, M.; Pourshohod, A.; Ghaffari, M.A.; Kheirollah, A.; Amin, M.; Zeinali, M.; Jamalan, M. Specific targeting of HER2-positive head and neck squamous cell carcinoma line hn5 by idarubicin-ZHER2 affibody conjugate. Curr. Cancer Drug Targets 2019, 19, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Sochaj-Gregorczyk, A.M.; Serwotka-Suszczak, A.M.; Otlewski, J. A novel affibody-auristatin e conjugate with a potent and selective activity against HER2+ cell lines. J. Immunother. 2016, 39, 223–232. [Google Scholar] [CrossRef]
- Strohl, W.R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half-lives of therapeutic proteins. J. Control. Release 2019, 301, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. Fcrn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Lawrence, P.B.; Price, J.L. How pegylation influences protein conformational stability. Curr. Opin. Chem. Biol. 2016, 34, 88–94. [Google Scholar] [CrossRef]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Su, W.; Zhang, W.; Wang, P.; Sattler, M.; Zou, P. Recent advances in half-life extension strategies for therapeutic peptides and proteins. Curr. Pharm. Des. 2018, 24, 4932–4946. [Google Scholar] [CrossRef]
- Jia, H.; Guo, Y.; Song, X.; Shao, C.; Wu, J.; Ma, J.; Shi, M.; Miao, Y.; Li, R.; Wang, D.; et al. Elimination of n-glycosylation by site mutation further prolongs the half-life of InF-α/Fc fusion proteins expressed in pichia pastoris. Microb. Cell Factories 2016, 15, 209. [Google Scholar] [CrossRef] [PubMed]
- Ulisse, S.; Baldini, E.; Sorrenti, S.; D’Armiento, M. The urokinase plasminogen activator system: A target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9, 32–71. [Google Scholar] [CrossRef]
- Janssen, E.M.; Dy, S.M.; Meara, A.S.; Kneuertz, P.J.; Presley, C.J.; Bridges, J.F. Analysis of patient preferences in lung cancer—Estimating acceptable tradeoffs between treatment benefit and side effects. Patient Prefer Adherence 2020, 14, 927–937. [Google Scholar] [CrossRef]
- Tincknell, G.; Piper, A.-K.; Aghmesheh, M.; Becker, T.; Vine, K.L.; Brungs, D.; Ranson, M. Experimental and clinical evidence supports the use of urokinase plasminogen activation system components as clinically relevant biomarkers in gastroesophageal adenocarcinoma. Cancers 2021, 13, 4097. [Google Scholar] [CrossRef]
- Lin, W.W.; Lu, Y.C.; Chuang, C.H.; and Cheng, T.L. Ab locks for improving the selectivity and safety of antibody drugs. J. Biomed. Sci. 2020, 27, 76. [Google Scholar] [CrossRef] [PubMed]
- Mazar, A.P. The urokinase plasminogen activator receptor (upar) as a target for the diagnosis and therapy of cancer. Anticancer Drugs 2001, 12, 387–400. [Google Scholar] [CrossRef]
- Orlova, A.; Magnusson, M.; Eriksson, T.L.J.; Nilsson, M.; Larsson, B.; Höidén-Guthenberg, I.; Widström, C.; Carlsson, J.; Tolmachev, V.; Ståhl, S.; et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006, 66, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Ståhl, S.; Uhlén, M.; Nygren, P.Å. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsén, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.; Uhlén, M. A synthetic igg-binding domain based on staphylococcal protein a. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.-Å.; Löfblom, J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Maderna, A.; Leverett, C.A. Recent advances in the development of new auristatins: Structural modifications and application in antibody drug conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Robles, V.; Malinao, M.-C.; Song, J.; Mendelsohn, B.A. Comparison of analytical methods for antibody-drug conjugates produced by chemical site-specific conjugation: First-generation ajicap. Anal. Chem. 2019, 91, 12724–12732. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-mediated protein ligation: A new method for protein engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef]
- van Kempen, L.C.; de Visser, K.E.; Coussens, L.M. Inflammation, proteases and cancer. Eur. J. Cancer 2006, 42, 728–734. [Google Scholar] [CrossRef]
- Boulware, K.T.; Daugherty, P.S. Protease specificity determination by using cellular libraries of peptide substrates (clips). Proc. Natl. Acad. Sci. USA 2006, 103, 7583–7588. [Google Scholar] [CrossRef] [PubMed]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.M.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 2013, 5, 207ra144. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, C.P.; Witte, M.D.; Theile, C.S.; Bozkurt, G.; Kundrat, L.; Blom, A.E.M.; Ploegh, H.L. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1787–1799. [Google Scholar] [CrossRef]
- Eigenbrot, C.; Ultsch, M.; Dubnovitsky, A.; Abrahmsén, L.; Härd, T. Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc. Natl. Acad. Sci. USA 2010, 107, 15039–15044. [Google Scholar] [CrossRef] [PubMed]
- Oroujeni, M.; Tano, H.; Vorobyeva, A.; Liu, Y.; Vorontsova, O.; Xu, T.; Westerlund, K.; Orlova, A.; Tolmachev, V.; Karlström, A.E. Affibody-mediated pna-based pretargeted cotreatment improves survival of trastuzumab-treated mice bearing her2-expressing xenografts. J. Nucl. Med. 2022, 63, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Guo, W.; Cao, L.; Liu, H.; Liu, J.; Xu, H.; Huang, W.; Wang, F.; Hong, Z. Fusion of an albumin-binding domain extends the half-life of immunotoxins. Int. J. Pharm. 2016, 511, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, S.; Orlova, A.; Wållberg, H.; Hansson, M.; Sandström, M.; Lewsley, R.; Wennborg, A.; Abrahmsén, L.; Tolmachev, V.; Feldwisch, J. Targeting of HER2-expressing tumors using 111in-ABY-025, a second-generation affibody molecule with a fundamentally reengineered scaffold. J. Nucl. Med. 2010, 51, 1131–1138. [Google Scholar] [CrossRef]
- Sandersjöö, L.; Jonsson, A.; Löfblom, J. A new prodrug form of affibody molecules (pro-affibody) is selectively activated by cancer-associated proteases. Cell. Mol. Life Sci. 2015, 72, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. Arx788, a site-specific anti-HER2 antibody-drug conjugate, demonstrates potent and selective activity in HER2-low and t-dm1-resistant breast and gastric cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Xu, Z.; Li, L.; Liu, W.; Dai, Z.; Zhao, Z.; Xiao, L.; Li, H.; Hu, C. Preclinical evaluation of MRG002, a novel HER2-targeting antibody-drug conjugate with potent antitumor activity against HER2-positive solid tumors. Antib. Ther. 2021, 4, 175–184. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dead/Total Mice | |
|---|---|
| Injected Dose (mg/kg) | Fc-U-ZHER2-MMAE |
| 23 | 0/5 |
| 45 | 0/5 |
| 90 | 0/5 |
| 180 | 2/5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Han, J.; Yang, L.; Cao, L.; Li, S.; Hong, Z. Tumor Site-Specific Cleavage Improves the Antitumor Efficacy of Antibody–Drug Conjugates. Int. J. Mol. Sci. 2023, 24, 11011. https://doi.org/10.3390/ijms241311011
Xu K, Han J, Yang L, Cao L, Li S, Hong Z. Tumor Site-Specific Cleavage Improves the Antitumor Efficacy of Antibody–Drug Conjugates. International Journal of Molecular Sciences. 2023; 24(13):11011. https://doi.org/10.3390/ijms241311011
Chicago/Turabian StyleXu, Keyuan, Jiani Han, Liu Yang, Li Cao, Shuang Li, and Zhangyong Hong. 2023. "Tumor Site-Specific Cleavage Improves the Antitumor Efficacy of Antibody–Drug Conjugates" International Journal of Molecular Sciences 24, no. 13: 11011. https://doi.org/10.3390/ijms241311011
APA StyleXu, K., Han, J., Yang, L., Cao, L., Li, S., & Hong, Z. (2023). Tumor Site-Specific Cleavage Improves the Antitumor Efficacy of Antibody–Drug Conjugates. International Journal of Molecular Sciences, 24(13), 11011. https://doi.org/10.3390/ijms241311011