

Targeting Opioid Receptors in Addiction and Drug Withdrawal: Where Are We Going?




Abstract
1. Introduction
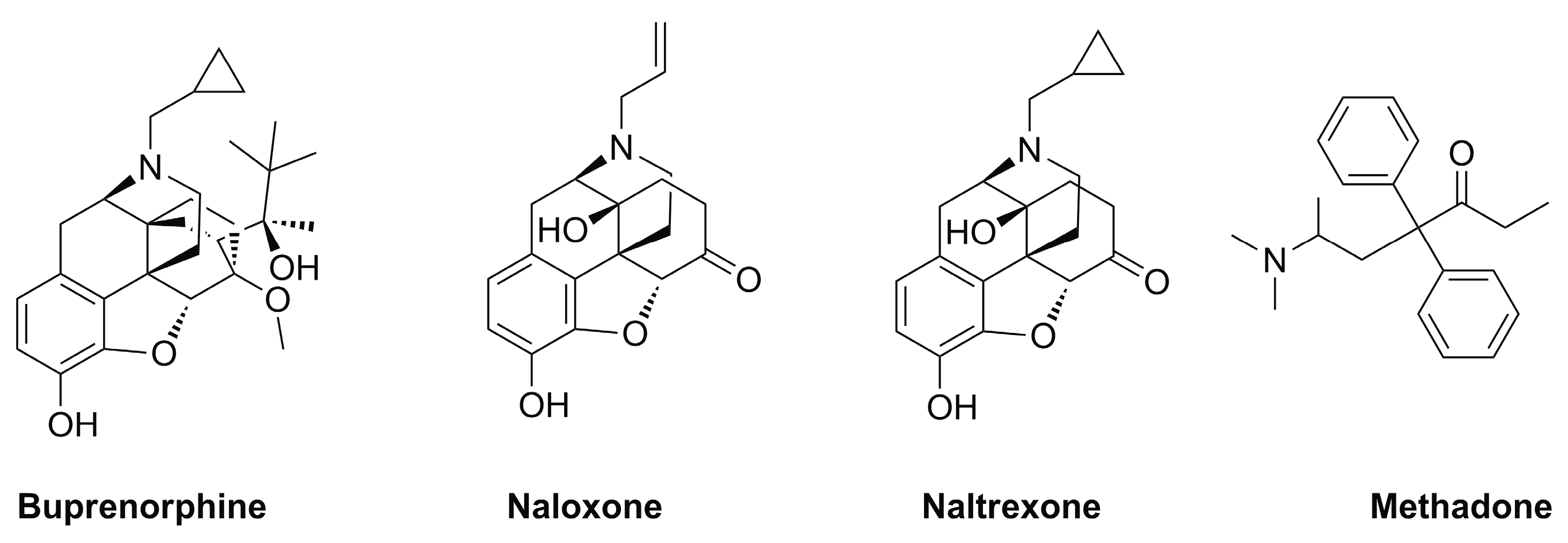
2. Opioid Receptor Functioning and Common Approved Drugs for Treating OUD
3. Psychosocial Interventions for Substance Abuse and Dependence
4. Mechanisms of Drug Withdrawal and Novel Therapeutic Options for Treating OUD
4.1. Promising Opioid Receptor Modulators for the Treatment and Prevention of OUD
4.1.1. Methocinnamox (MCAM)
4.1.2. Mitragynine (Kratom)
4.2. Safer and More Tolerable Therapeutics for Treating Severe Pain: Oliceridine, the Opioid of the 21st Century?
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azadfard, M.; Huecker, M.R.; Leaming, J.M. Opioid Addiction [Updated 1 January 2023]; StatPearls: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448203/ (accessed on 10 January 2023).
- Available online: https://www.cdc.gov/opioids/healthcare-professionals/prescribing/opioid-use-disorder.html (accessed on 10 January 2023).
- Strang, J.; Volkow, N.D.; Degenhardt, L.; Hickman, M.; Johnson, K.; Koob, G.F.; Marshall, B.D.L.; Tyndall, M.; Walsh, S.L. Opioid use disorder. Nat. Rev. Dis. Prim. 2020, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Korthuis, P.T.; McCarty, D.; Weimer, M.; Bougatsos, C.; Blazina, I.; Zakher, B.; Grusing, S.; Devine, B.; Chou, R. Primary Care-Based Models for the Treatment of Opioid Use Disorder: A Scoping Review. Ann. Intern. Med. 2017, 166, 268–278. [Google Scholar] [CrossRef]
- Donroe, J.H.; Bhatraju, E.P.; Tsui, J.I.; Edelman, E.J. Identification and Management of Opioid Use Disorder in Primary Care: An Update. Curr. Psychiatry Rep. 2020, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, K.A.; Ponce Terashima, J.; McCarty, D. Opioid use disorder and treatment: Challenges and opportunities. BMC Health Serv. Res. 2019, 19, 884. [Google Scholar] [CrossRef] [PubMed]
- Busse, J.W.; Wang, L.; Kamaleldin, M.; Craigie, S.; Riva, J.J.; Montoya, L.; Mulla, S.M.; Lopes, L.C.; Vogel, N.; Chen, E.; et al. Opioids for Chronic Noncancer Pain: A Systematic Review and Meta-analysis. J. Am. Med. Assoc. 2018, 320, 2448–2460. [Google Scholar] [CrossRef]
- Wiffen, P.J.; Wee, B.; Derry, S.; Bell, R.F.; Moore, R.A. Opioids for cancer pain—An overview of Cochrane reviews. Cochrane Database Syst. Rev. 2017, 7, CD012592. [Google Scholar] [CrossRef]
- Quigley, C. Opioids in people with cancer-related pain. BMJ Clin. Evid. 2008, 2008, 2408. [Google Scholar]
- Busse, J.W.; Craigie, S.; Juurlink, D.N.; Buckley, D.N.; Wang, L.; Couban, R.J.; Agoritsas, T.; Akl, E.A.; Carrasco-Labra, A.; Cooper, L.; et al. Guideline for opioid therapy and chronic noncancer pain. Can. Med. Assoc. J. 2017, 189, E659–E666. [Google Scholar] [CrossRef]
- Nadeau, S.E.; Lawhern, R.A. Management of chronic non-cancer pain: A framework. Pain Manag. 2022, 12, 751–777. [Google Scholar] [CrossRef]
- Bertin, C.; Delage, N.; Rolland, B.; Pennel, L.; Fatseas, M.; Trouvin, A.P.; Delorme, J.; Chenaf, C.; Authier, N. Analgesic opioid use disorders in patients with chronic non-cancer pain: A holistic approach for tailored management. Neurosci. Biobehav. Rev. 2021, 121, 160–174. [Google Scholar] [CrossRef]
- Graven-Nielsen, C.S.; Knoph, C.S.; Okdahl, T.; Hoyer, K.L.; Krogh, K.; Hellstrom, P.M.; Drewes, A.M. Opioids in the Treatment of Chronic Idiopathic Diarrhea in Humans-A Systematic Review and Treatment Guideline. J. Clin. Med. 2023, 12, 2488. [Google Scholar] [CrossRef] [PubMed]
- Schiller, L.R.; Pardi, D.S.; Sellin, J.H. Chronic Diarrhea: Diagnosis and Management. Clin. Gastroenterol. Hepatol. 2017, 15, 182–193.e13. [Google Scholar] [CrossRef] [PubMed]
- Ruppin, H. Review: Loperamide—A potent antidiarrhoeal drug with actions along the alimentary tract. Aliment. Pharmacol. Ther. 1987, 1, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Belvisi, M.G.; Geppetti, P. Cough. 7: Current and future drugs for the treatment of chronic cough. Thorax 2004, 59, 438–440. [Google Scholar] [CrossRef] [PubMed]
- On, P.C. Updates in treatment of adults with chronic cough. Am. J. Manag. Care 2020, 26, S239–S245. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, F.; Maffulli, N.; Baroncini, A.; Eschweiler, J.; Tingart, M.; Quack, V. Opioids for chronic low back pain management: A Bayesian network meta-analysis. Expert. Rev. Clin. Pharmacol. 2021, 14, 635–641. [Google Scholar] [CrossRef]
- Petzke, F.; Klose, P.; Welsch, P.; Sommer, C.; Hauser, W. Opioids for chronic low back pain: An updated systematic review and meta-analysis of efficacy, tolerability and safety in randomized placebo-controlled studies of at least 4 weeks of double-blind duration. Eur. J. Pain 2020, 24, 497–517. [Google Scholar] [CrossRef]
- Derakhshan, I. The diagnosis and treatment of chronic migraine: The case for daily scheduled opioid treatment in chronic headache. Ther. Adv. Chronic. Dis. 2015, 6, 389. [Google Scholar] [CrossRef]
- Rothrock, J.F. Opiate and opioid (“narcotic”) therapy for acute migraine headache. Headache 2010, 50, 1255–1256. [Google Scholar] [CrossRef]
- Srivastava, D.; Hill, S.; Carty, S.; Rockett, M.; Bastable, R.; Knaggs, R.; Lambert, D.; Levy, N.; Hughes, J.; Wilkinson, P. Surgery and opioids: Evidence-based expert consensus guidelines on the perioperative use of opioids in the United Kingdom. Br. J. Anaesth. 2021, 126, 1208–1216. [Google Scholar] [CrossRef]
- Kelley-Quon, L.I.; Kirkpatrick, M.G.; Ricca, R.L.; Baird, R.; Harbaugh, C.M.; Brady, A.; Garrett, P.; Wills, H.; Argo, J.; Diefenbach, K.A.; et al. Guidelines for Opioid Prescribing in Children and Adolescents After Surgery: An Expert Panel Opinion. JAMA Surg. 2021, 156, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Myles, P.S.; Bui, T. Opioid-free analgesia after surgery. Lancet 2022, 399, 2245–2247. [Google Scholar] [CrossRef] [PubMed]
- Berecki-Gisolf, J.; Hassani-Mahmooei, B.; Collie, A.; McClure, R. Prescription Opioid and Benzodiazepine Use After Road Traffic Injury. Pain Med. 2016, 17, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Bemand-Qureshi, L.; Gishen, F.; Tookman, A. Opioid use in palliative care: New developments and guidelines. Prescriber 2019, 30, 25–31. [Google Scholar] [CrossRef]
- Lau, J.; Mazzotta, P.; Whelan, C.; Abdelaal, M.; Clarke, H.; Furlan, A.D.; Smith, A.; Husain, A.; Fainsinger, R.; Hui, D.; et al. Opioid safety recommendations in adult palliative medicine: A North American Delphi expert consensus. BMJ Support. Palliat. Care 2022, 12, 81–90. [Google Scholar] [CrossRef]
- Rome, R.B.; Luminais, H.H.; Bourgeois, D.A.; Blais, C.M. The role of palliative care at the end of life. Ochsner. J. 2011, 11, 348–352. [Google Scholar] [PubMed]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, S105–S120. [Google Scholar] [CrossRef]
- Adverse effects of opioids for non-cancer pain. Drug Ther. Bull. 2018, 56, 15–16. [CrossRef]
- Els, C.; Jackson, T.D.; Kunyk, D.; Lappi, V.G.; Sonnenberg, B.; Hagtvedt, R.; Sharma, S.; Kolahdooz, F.; Straube, S. Adverse events associated with medium- and long-term use of opioids for chronic non-cancer pain: An overview of Cochrane Reviews. Cochrane Database Syst. Rev. 2017, 10, CD012509. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, S. Opioid Analgesics Adverse Effects: The Other Side of the Coin. Curr. Pharm. Des. 2019, 25, 3197–3202. [Google Scholar] [CrossRef]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef] [PubMed]
- Corli, O.; Santucci, C.; Corsi, N.; Radrezza, S.; Galli, F.; Bosetti, C. The Burden of Opioid Adverse Events and the Influence on Cancer Patients’ Symptomatology. J. Pain Symptom Manag. 2019, 57, 899–908.e6. [Google Scholar] [CrossRef] [PubMed]
- Schug, S.A.; Zech, D.; Grond, S. Adverse effects of systemic opioid analgesics. Drug Saf. 1992, 7, 200–213. [Google Scholar] [CrossRef]
- Vella-Brincat, J.; MacLeod, A.D. Adverse Effects of Opioids on the Central Nervous Systems of Palliative Care Patients. J. Pain Palliat. Care Pharmacother. 2009, 21, 15–25. [Google Scholar] [CrossRef]
- Pergolizzi, J.V.; Varrassi, G.; Paladini, A.; LeQuang, J. Stopping or Decreasing Opioid Therapy in Patients on Chronic Opioid Therapy. Pain Ther. 2019, 8, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Vallersnes, O.M.; Jacobsen, D.; Ekeberg, O.; Brekke, M. Mortality, morbidity and follow-up after acute poisoning by substances of abuse: A prospective observational cohort study. Scand. J. Public Health 2019, 47, 452–461. [Google Scholar] [CrossRef]
- Kosten, T.R.; Baxter, L.E. Review article: Effective management of opioid withdrawal symptoms: A gateway to opioid dependence treatment. Am. J. Addict. 2019, 28, 55–62. [Google Scholar] [CrossRef]
- Pergolizzi, J.V., Jr.; Raffa, R.B.; Rosenblatt, M.H. Opioid withdrawal symptoms, a consequence of chronic opioid use and opioid use disorder: Current understanding and approaches to management. J. Clin. Pharm. Ther. 2020, 45, 892–903. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- Gaskin, D.J.; Richard, P. The economic costs of pain in the United States. J. Pain 2012, 13, 715–724. [Google Scholar] [CrossRef]
- Florence, C.; Luo, F.; Rice, K. The economic burden of opioid use disorder and fatal opioid overdose in the United States, 2017. Drug Alcohol. Depend. 2021, 218, 108350. [Google Scholar] [CrossRef]
- Smith, T.J.; Hillner, B.E. The Cost of Pain. JAMA Netw. Open 2019, 2, e191532. [Google Scholar] [CrossRef] [PubMed]
- Machelska, H.; Celik, M.O. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.H.; Pasternak, G.W. Historical review: Opioid receptors. Trends Pharmacol. Sci. 2003, 24, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The delta-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Toya, T.; Katao, Y.; Maekawa, K.; Nakamura, S.; Onogi, T.; Kaneko, S.; Satoh, M. Cloning and expression of a cDNA for the rat kappa-opioid receptor. FEBS Lett. 1993, 329, 291–295. [Google Scholar] [CrossRef]
- Cox, B.M. A Concise Review of Concepts in Opioid Pharmacology up to the Discovery of Endogenous Opioids. Mol. Pharmacol. 2020, 98, 392–400. [Google Scholar] [CrossRef]
- Higginbotham, J.A.; Markovic, T.; Massaly, N.; Moron, J.A. Endogenous opioid systems alterations in pain and opioid use disorder. Front. Syst. Neurosci. 2022, 16, 1014768. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Tafi, A.; Desaubry, L.; Nebigil, C.G. Discovery of GPCR ligands for probing signal transduction pathways. Front. Pharmacol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Yamada, D.; Takahashi, J.; Iio, K.; Nagase, H.; Saitoh, A. Modulation of glutamatergic synaptic transmission and neuronal excitability in the prelimbic medial prefrontal cortex via delta-opioid receptors in mice. Biochem. Biophys. Res. Commun. 2021, 560, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Freye, E.; Levy, J. Constitutive opioid receptor activation: A prerequisite mechanism involved in acute opioid withdrawal. Addict. Biol. 2005, 10, 131–137. [Google Scholar] [CrossRef]
- Trescot, A.M.; Datta, S.; Lee, M.; Hansen, H. Opioid pharmacology. Pain Physician 2008, 11, S133–S153. [Google Scholar] [CrossRef]
- Le Merrer, J.; Becker, J.A.; Befort, K.; Kieffer, B.L. Reward processing by the opioid system in the brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef]
- Reeves, K.C.; Shah, N.; Munoz, B.; Atwood, B.K. Opioid Receptor-Mediated Regulation of Neurotransmission in the Brain. Front. Mol. Neurosci. 2022, 15, 919773. [Google Scholar] [CrossRef]
- Eisenstein, T.K. The Role of Opioid Receptors in Immune System Function. Front. Immunol. 2019, 10, 2904. [Google Scholar] [CrossRef]
- Patel, D.; Callaway, J.; Vaezi, M. Opioid-Induced Foregut Dysfunction. Am. J. Gastroenterol. 2019, 114, 1716–1725. [Google Scholar] [CrossRef]
- Drews, E.; Zimmer, A. Modulation of alcohol and nicotine responses through the endogenous opioid system. Prog. Neurobiol. 2010, 90, 1–15. [Google Scholar] [CrossRef]
- Galaj, E.; Xi, Z.X. Progress in opioid reward research: From a canonical two-neuron hypothesis to two neural circuits. Pharmacol. Biochem. Behav. 2021, 200, 173072. [Google Scholar] [CrossRef] [PubMed]
- Reisine, T.; Bell, G.I. Molecular biology of opioid receptors. Trends Neurosci. 1993, 16, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Jarvie, B.C.; Robinson, B.G.; Hentges, S.T.; Williams, J.T. Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron 2014, 82, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Dumas, E.O.; Pollack, G.M. Opioid tolerance development: A pharmacokinetic/pharmacodynamic perspective. AAPS J. 2008, 10, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ma, R.; Jin, Y.; Fang, J.; Du, J.; Shao, X.; Liang, Y.; Fang, J. Molecular mechanisms of opioid tolerance: From opioid receptors to inflammatory mediators (Review). Exp. Ther. Med. 2021, 22, 1004. [Google Scholar] [CrossRef] [PubMed]
- Eidson, L.N.; Murphy, A.Z. Inflammatory mediators of opioid tolerance: Implications for dependency and addiction. Peptides 2019, 115, 51–58. [Google Scholar] [CrossRef]
- Kosten, T.R.; George, T.P. The neurobiology of opioid dependence: Implications for treatment. Sci. Pract. Perspect. 2002, 1, 13–20. [Google Scholar] [CrossRef]
- Bassareo, V.; Tanda, G.; Di Chiara, G. Increase of extracellular dopamine in the medial prefrontal cortex during spontaneous and naloxone-precipitated opiate abstinence. Psychopharmacology 1995, 122, 202–205. [Google Scholar] [CrossRef]
- Rossetti, Z.L.; Longu, G.; Mercuro, G.; Gessa, G.L. Extraneuronal noradrenaline in the prefrontal cortex of morphine-dependent rats: Tolerance and withdrawal mechanisms. Brain Res. 1993, 609, 316–320. [Google Scholar] [CrossRef]
- Espejo, E.F.; Serrano, M.I.; Caille, S.; Stinus, L. Behavioral expression of opiate withdrawal is altered after prefrontocortical dopamine depletion in rats: Monoaminergic correlates. Neuropsychopharmacology 2001, 25, 204–212. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, H.L.; Xiang, X.H.; Zhao, Y. The role of glutamate and its receptors in mesocorticolimbic dopaminergic regions in opioid addiction. Neurosci. Biobehav. Rev. 2009, 33, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K. Afferent effects on locus coeruleus in opiate withdrawal. Prog. Brain Res. 1991, 88, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Leri, F.; Ho, A.; Kreek, M.J. Suppression of hypothalamic-pituitary-adrenal axis by acute heroin challenge in rats during acute and chronic withdrawal from chronic heroin administration. Neurochem. Res. 2013, 38, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Shokri-Kojori, E.; Wang, G.J.; Volkow, N.D. Naloxone precipitated withdrawal increases dopamine release in the dorsal striatum of opioid dependent men. Transl. Psychiatry 2021, 11, 445. [Google Scholar] [CrossRef] [PubMed]
- Coffin, P.O.; Behar, E.; Rowe, C.; Santos, G.M.; Coffa, D.; Bald, M.; Vittinghoff, E. Nonrandomized Intervention Study of Naloxone Coprescription for Primary Care Patients Receiving Long-Term Opioid Therapy for Pain. Ann. Intern. Med. 2016, 165, 245–252. [Google Scholar] [CrossRef]
- Kleber, H.D. Pharmacologic treatments for opioid dependence: Detoxification and maintenance options. Dialogues Clin. Neurosci. 2007, 9, 455–470. [Google Scholar] [CrossRef]
- Dahan, A.; Yassen, A.; Romberg, R.; Sarton, E.; Teppema, L.; Olofsen, E.; Danhof, M. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br. J. Anaesth. 2006, 96, 627–632. [Google Scholar] [CrossRef]
- Connery, H.S. Medication-assisted treatment of opioid use disorder: Review of the evidence and future directions. Harv. Rev. Psychiatry 2015, 23, 63–75. [Google Scholar] [CrossRef]
- Fiellin, D.A.; Schottenfeld, R.S.; Cutter, C.J.; Moore, B.A.; Barry, D.T.; O’Connor, P.G. Primary care-based buprenorphine taper vs maintenance therapy for prescription opioid dependence: A randomized clinical trial. JAMA Intern. Med. 2014, 174, 1947–1954. [Google Scholar] [CrossRef]
- Kakko, J.; Svanborg, K.D.; Kreek, M.J.; Heilig, M. 1-year retention and social function after buprenorphine-assisted relapse prevention treatment for heroin dependence in Sweden: A randomised, placebo-controlled trial. Lancet 2003, 361, 662–668. [Google Scholar] [CrossRef]
- Mattick, R.P.; Breen, C.; Kimber, J.; Davoli, M. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst. Rev. 2014, 6, CD002207. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.; Lamb, K.; Thomas, M.L.; Khentigan, W. Buprenorphine Maintenance Treatment of Opiate Dependence: Correlations Between Prescriber Beliefs and Practices. Subst. Use Misuse 2016, 51, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kreek, M.J. Methadone-related opioid agonist pharmacotherapy for heroin addiction. History, recent molecular and neurochemical research and future in mainstream medicine. Ann. N. Y. Acad. Sci. 2000, 909, 186–216. [Google Scholar] [CrossRef] [PubMed]
- Koehl, J.L.; Zimmerman, D.E.; Bridgeman, P.J. Medications for management of opioid use disorder. Am. J. Health Syst. Pharm. 2019, 76, 1097–1103. [Google Scholar] [CrossRef]
- Mattick, R.P.; Breen, C.; Kimber, J.; Davoli, M. Methadone maintenance therapy versus no opioid replacement therapy for opioid dependence. Cochrane Database Syst. Rev. 2009, 2009, CD002209. [Google Scholar] [CrossRef]
- Schwartz, R.P.; Highfield, D.A.; Jaffe, J.H.; Brady, J.V.; Butler, C.B.; Rouse, C.O.; Callaman, J.M.; O’Grady, K.E.; Battjes, R.J. A randomized controlled trial of interim methadone maintenance. Arch. Gen. Psychiatry 2006, 63, 102–109. [Google Scholar] [CrossRef]
- Krupitsky, E.; Nunes, E.V.; Ling, W.; Illeperuma, A.; Gastfriend, D.R.; Silverman, B.L. Injectable extended-release naltrexone for opioid dependence: A double-blind, placebo-controlled, multicentre randomised trial. Lancet 2011, 377, 1506–1513. [Google Scholar] [CrossRef]
- Lee, J.D.; Nunes, E.V., Jr.; Novo, P.; Bachrach, K.; Bailey, G.L.; Bhatt, S.; Farkas, S.; Fishman, M.; Gauthier, P.; Hodgkins, C.C.; et al. Comparative effectiveness of extended-release naltrexone versus buprenorphine-naloxone for opioid relapse prevention (X:BOT): A multicentre, open-label, randomised controlled trial. Lancet 2018, 391, 309–318. [Google Scholar] [CrossRef]
- Nunes, E.V.; Krupitsky, E.; Ling, W.; Zummo, J.; Memisoglu, A.; Silverman, B.L.; Gastfriend, D.R. Treating Opioid Dependence With Injectable Extended-Release Naltrexone (XR-NTX): Who Will Respond? J. Addict. Med. 2015, 9, 238–243. [Google Scholar] [CrossRef]
- Minozzi, S.; Amato, L.; Vecchi, S.; Davoli, M.; Kirchmayer, U.; Verster, A. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst. Rev. 2011, 2011, CD001333. [Google Scholar] [CrossRef]
- Krupitsky, E.; Nunes, E.V.; Ling, W.; Gastfriend, D.R.; Memisoglu, A.; Silverman, B.L. Injectable extended-release naltrexone (XR-NTX) for opioid dependence: Long-term safety and effectiveness. Addiction 2013, 108, 1628–1637. [Google Scholar] [CrossRef]
- Syed, Y.Y.; Keating, G.M. Extended-release intramuscular naltrexone (VIVITROL(R)): A review of its use in the prevention of relapse to opioid dependence in detoxified patients. CNS Drugs 2013, 27, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.; Mandell, K.; Johnson, K.; Chatterjee, D.; Vanness, D.J. Cost-Effectiveness of Injectable Extended-Release Naltrexone Compared with Methadone Maintenance and Buprenorphine Maintenance Treatment for Opioid Dependence. Subst. Abus. 2023, 36, 226–231. [Google Scholar] [CrossRef]
- Available online: https://nida.nih.gov/publications/research-reports/medications-to-treat-opioid-addiction/efficacy-medications-opioid-use-disorder (accessed on 10 June 2023).
- Sordo, L.; Barrio, G.; Bravo, M.J.; Indave, B.I.; Degenhardt, L.; Wiessing, L.; Ferri, M.; Pastor-Barriuso, R. Mortality risk during and after opioid substitution treatment: Systematic review and meta-analysis of cohort studies. Br. Med. J. 2017, 357, j1550. [Google Scholar] [CrossRef]
- Fareed, A.; Patil, D.; Scheinberg, K.; Blackinton Gale, R.; Vayalapalli, S.; Casarella, J.; Drexler, K. Comparison of QTc interval prolongation for patients in methadone versus buprenorphine maintenance treatment: A 5-year follow-up. J. Addict. Dis. 2013, 32, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Berger, O.; Rector, K.; Meredith, J.; Sebaaly, J. Evaluation of drug-drug interactions in hospitalized patients on medications for OUD. Ment. Health Clin. 2021, 11, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Schuckit, M.A. Treatment of Opioid-Use Disorders. N. Engl. J. Med. 2016, 375, 357–368. [Google Scholar] [CrossRef]
- Blanco, C.; Volkow, N.D. Management of opioid use disorder in the USA: Present status and future directions. Lancet 2019, 393, 1760–1772. [Google Scholar] [CrossRef]
- Volkow, N.D.; Jones, E.B.; Einstein, E.B.; Wargo, E.M. Prevention and Treatment of Opioid Misuse and Addiction: A Review. JAMA Psychiatry 2019, 76, 208–216. [Google Scholar] [CrossRef]
- Marsden, J.; Stillwell, G.; Hellier, J.; Brown, A.M.; Byford, S.; Kelleher, M.; Kelly, J.; Murphy, C.; Shearer, J.; Mitcheson, L. Effectiveness of adjunctive, personalised psychosocial intervention for non-response to opioid agonist treatment: Study protocol for a pragmatic randomised controlled trial. Contemp. Clin. Trials 2017, 53, 36–43. [Google Scholar] [CrossRef]
- McHugh, R.K.; Hearon, B.A.; Otto, M.W. Cognitive behavioral therapy for substance use disorders. Psychiatr Clin. N. Am. 2010, 33, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Kakko, J.; Alho, H.; Baldacchino, A.; Molina, R.; Nava, F.A.; Shaya, G. Craving in Opioid Use Disorder: From Neurobiology to Clinical Practice. Front. Psychiatry 2019, 10, 592. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.C.; Murphy, J.G.; Witkiewitz, K.; Hand, S.B.; Thomas, F.; Johnson, K.C.; Cowan, R.; Harris, M.; Derefinko, K.J. Use of a sequential multiple assignment randomized trial to test contingency management and an integrated behavioral economic and mindfulness intervention for buprenorphine-naloxone medication adherence for opioid use disorder. Trials 2023, 24, 237. [Google Scholar] [CrossRef]
- Petry, N.M.; Martin, B. Low-cost contingency management for treating cocaine- and opioid-abusing methadone patients. J. Consult. Clin. Psychol. 2002, 70, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, M.; Podus, D.; Finney, J.; Greenwell, L.; Roll, J. Contingency management for treatment of substance use disorders: A meta-analysis. Addiction 2006, 101, 1546–1560. [Google Scholar] [CrossRef]
- Messina, N.; Farabee, D.; Rawson, R. Treatment responsivity of cocaine-dependent patients with antisocial personality disorder to cognitive-behavioral and contingency management interventions. J. Consult. Clin. Psychol. 2003, 71, 320–329. [Google Scholar] [CrossRef]
- Preston, K.L.; Umbricht, A.; Epstein, D.H. Abstinence reinforcement maintenance contingency and one-year follow-up. Drug Alcohol. Depend. 2002, 67, 125–137. [Google Scholar] [CrossRef]
- Willner-Reid, J.; Whitaker, D.; Epstein, D.H.; Phillips, K.A.; Pulaski, A.R.; Preston, K.L.; Willner, P. Cognitive-behavioural therapy for heroin and cocaine use: Ecological momentary assessment of homework simplification and compliance. Psychol. Psychother 2016, 89, 276–293. [Google Scholar] [CrossRef]
- Dutra, L.; Stathopoulou, G.; Basden, S.L.; Leyro, T.M.; Powers, M.B.; Otto, M.W. A meta-analytic review of psychosocial interventions for substance use disorders. Am. J. Psychiatry 2008, 165, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bowen, S.; Chawla, N.; Collins, S.E.; Witkiewitz, K.; Hsu, S.; Grow, J.; Clifasefi, S.; Garner, M.; Douglass, A.; Larimer, M.E.; et al. Mindfulness-based relapse prevention for substance use disorders: A pilot efficacy trial. Subst. Abus. 2009, 30, 295–305. [Google Scholar] [CrossRef]
- Price, C.J.; Merrill, J.O.; McCarty, R.L.; Pike, K.C.; Tsui, J.I. A pilot study of mindful body awareness training as an adjunct to office-based medication treatment of opioid use disorder. J. Subst. Abus. Treat 2020, 108, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.T.; Killeen, T.; Baker, N.L. Efficacy of mindfulness-based relapse prevention in veterans with substance use disorders: Design and methodology of a randomized clinical trial. Contemp. Clin. Trials 2021, 105, 106393. [Google Scholar] [CrossRef] [PubMed]
- Garland, E.L.; Hanley, A.W.; Kline, A.; Cooperman, N.A. Mindfulness-Oriented Recovery Enhancement reduces opioid craving among individuals with opioid use disorder and chronic pain in medication assisted treatment: Ecological momentary assessments from a stage 1 randomized controlled trial. Drug Alcohol. Depend. 2019, 203, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.F.; Kaminer, Y.; Kahler, C.W.; Hoeppner, B.; Yeterian, J.; Cristello, J.V.; Timko, C. A pilot randomized clinical trial testing integrated 12-Step facilitation (iTSF) treatment for adolescent substance use disorder. Addiction 2017, 112, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Attwood, L.O.; McKechnie, M.; Vujovic, O.; Higgs, P.; Lloyd-Jones, M.; Doyle, J.S.; Stewardson, A.J. Review of management priorities for invasive infections in people who inject drugs: Highlighting the need for patient-centred multidisciplinary care. Med. J. Aust. 2022, 217, 102–109. [Google Scholar] [CrossRef]
- Levengood, T.W.; Yoon, G.H.; Davoust, M.J.; Ogden, S.N.; Marshall, B.D.L.; Cahill, S.R.; Bazzi, A.R. Supervised Injection Facilities as Harm Reduction: A Systematic Review. Am. J. Prev. Med. 2021, 61, 738–749. [Google Scholar] [CrossRef]
- Giulini, F.; Keenan, E.; Killeen, N.; Ivers, J.H. A Systematized Review of Drug-checking and Related Considerations for Implementation as A Harm Reduction Intervention. J. Psychoact. Drugs 2023, 55, 85–93. [Google Scholar] [CrossRef]
- Lloyd-Smith, E.; Wood, E.; Zhang, R.; Tyndall, M.W.; Sheps, S.; Montaner, J.S.; Kerr, T. Determinants of hospitalization for a cutaneous injection-related infection among injection drug users: A cohort study. BMC Public Health 2010, 10, 327. [Google Scholar] [CrossRef]
- Madah-Amiri, D.; Skulberg, A.K.; Braarud, A.C.; Dale, O.; Heyerdahl, F.; Lobmaier, P.; Clausen, T. Ambulance-attended opioid overdoses: An examination into overdose locations and the role of a safe injection facility. Subst. Abus. 2019, 40, 383–388. [Google Scholar] [CrossRef]
- Marshall, B.D.; Milloy, M.J.; Wood, E.; Montaner, J.S.; Kerr, T. Reduction in overdose mortality after the opening of North America’s first medically supervised safer injecting facility: A retrospective population-based study. Lancet 2011, 377, 1429–1437. [Google Scholar] [CrossRef]
- Myer, A.J.; Belisle, L. Highs and Lows: An Interrupted Time-Series Evaluation of the Impact of North America’s Only Supervised Injection Facility on Crime. J. Drug Issues 2017, 48, 36–49. [Google Scholar] [CrossRef]
- Salmon, A.M.; van Beek, I.; Amin, J.; Kaldor, J.; Maher, L. The impact of a supervised injecting facility on ambulance call-outs in Sydney, Australia. Addiction 2010, 105, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, J.A.; Wood, E.; Small, W.; Li, K.; Tyndall, M.; Montaner, J.; Kerr, T. Changes in injecting practices associated with the use of a medically supervised safer injection facility. J. Public Health 2007, 29, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Gold, M.S.; Fuehrlein, B.S. Looking beyond the opioid receptor: A desperate need for new treatments for opioid use disorder. J. Neurol. Sci. 2022, 432, 120094. [Google Scholar] [CrossRef] [PubMed]
- Varadi, A.; Marrone, G.F.; Palmer, T.C.; Narayan, A.; Szabo, M.R.; Le Rouzic, V.; Grinnell, S.G.; Subrath, J.J.; Warner, E.; Kalra, S.; et al. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit beta-Arrestin-2. J. Med. Chem. 2016, 59, 8381–8397. [Google Scholar] [CrossRef]
- Torres-Lockhart, K.E.; Lu, T.Y.; Weimer, M.B.; Stein, M.R.; Cunningham, C.O. Clinical Management of Opioid Withdrawal. Addiction 2022, 117, 2540–2550. [Google Scholar] [CrossRef]
- Jordan, C.G.; Kennalley, A.L.; Roberts, A.L.; Nemes, K.M.; Dolma, T.; Piper, B.J. The Potential of Methocinnamox as a Future Treatment for Opioid Use Disorder: A Narrative Review. Pharmacy 2022, 10, 48. [Google Scholar] [CrossRef]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef]
- De Aquino, J.P.; Bahji, A.; Gomez, O.; Sofuoglu, M. Alleviation of opioid withdrawal by cannabis and delta-9-tetrahydrocannabinol: A systematic review of observational and experimental human studies. Drug Alcohol. Depend. 2022, 241, 109702. [Google Scholar] [CrossRef]
- Wang, S.C.; Chen, Y.C.; Lee, C.H.; Cheng, C.M. Opioid Addiction, Genetic Susceptibility, and Medical Treatments: A Review. Int. J. Mol. Sci. 2019, 20, 4294. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, C.A.; Kearney-Ramos, T.; Dowdle, L.T.; Hamilton, S.; DeVries, W.; Mithoefer, O.; Austelle, C.; Lench, D.H.; Correia, B.; Canterberry, M.; et al. Developing Repetitive Transcranial Magnetic Stimulation (rTMS) as a Treatment Tool for Cocaine Use Disorder: A Series of Six Translational Studies. Curr. Behav. Neurosci. Rep. 2017, 4, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Razza, L.B.; Palumbo, P.; Moffa, A.H.; Carvalho, A.F.; Solmi, M.; Loo, C.K.; Brunoni, A.R. A systematic review and meta-analysis on the effects of transcranial direct current stimulation in depressive episodes. Depress. Anxiety 2020, 37, 594–608. [Google Scholar] [CrossRef]
- Troster, A.I. Neuropsychology of deep brain stimulation in neurology and psychiatry. Front. Biosci. 2009, 14, 1857–1879. [Google Scholar] [CrossRef]
- Bauer, S.; Baier, H.; Baumgartner, C.; Bohlmann, K.; Fauser, S.; Graf, W.; Hillenbrand, B.; Hirsch, M.; Last, C.; Lerche, H.; et al. Transcutaneous Vagus Nerve Stimulation (tVNS) for Treatment of Drug-Resistant Epilepsy: A Randomized, Double-Blind Clinical Trial (cMPsE02). Brain Stimul. 2016, 9, 356–363. [Google Scholar] [CrossRef]
- Bond, A.E.; Shah, B.B.; Huss, D.S.; Dallapiazza, R.F.; Warren, A.; Harrison, M.B.; Sperling, S.A.; Wang, X.Q.; Gwinn, R.; Witt, J.; et al. Safety and Efficacy of Focused Ultrasound Thalamotomy for Patients With Medication-Refractory, Tremor-Dominant Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2017, 74, 1412–1418. [Google Scholar] [CrossRef]
- Mahoney, J.J., 3rd; Hanlon, C.A.; Marshalek, P.J.; Rezai, A.R.; Krinke, L. Transcranial magnetic stimulation, deep brain stimulation, and other forms of neuromodulation for substance use disorders: Review of modalities and implications for treatment. J. Neurol. Sci. 2020, 418, 117149. [Google Scholar] [CrossRef]
- Bogenschutz, M.P.; Johnson, M.W. Classic hallucinogens in the treatment of addictions. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 250–258. [Google Scholar] [CrossRef]
- Shao, W.; Kuhn, C.; Mayr, D.; Ditsch, N.; Kailuwait, M.; Wolf, V.; Harbeck, N.; Mahner, S.; Jeschke, U.; Cavailles, V.; et al. Cytoplasmic LXR expression is an independent marker of poor prognosis for patients with early stage primary breast cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 2535–2544. [Google Scholar] [CrossRef]
- Rodrigues, L.S.; Rossi, G.N.; Rocha, J.M.; Osório, F.L.; Bouso, J.C.; Hallak, J.E.C.; Dos Santos, R.G. Effects of ayahuasca and its alkaloids on substance use disorders: An updated (2016–2020) systematic review of preclinical and human studies. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 541–556. [Google Scholar] [CrossRef]
- Smith, A.C.W.; Kenny, P.J. MicroRNAs regulate synaptic plasticity underlying drug addiction. Genes Brain Behav. 2018, 17, e12424. [Google Scholar] [CrossRef] [PubMed]
- Vasiliu, O. Current Trends and Perspectives in the Immune Therapy for Substance Use Disorders. Front. Psychiatry 2022, 13, 882491. [Google Scholar] [CrossRef] [PubMed]
- Bremer, P.T.; Janda, K.D. Conjugate Vaccine Immunotherapy for Substance Use Disorder. Pharmacol. Rev. 2017, 69, 298–315. [Google Scholar] [CrossRef]
- Zamora, J.C.; Smith, H.R.; Jennings, E.M.; Chavera, T.S.; Kotipalli, V.; Jay, A.; Husbands, S.M.; Disney, A.; Berg, K.A.; Clarke, W.P. Long-term antagonism and allosteric regulation of mu opioid receptors by the novel ligand, methocinnamox. Pharmacol. Res. Perspect. 2021, 9, e00887. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.A. The lasting impact of methocinnamox on opioid self-administration. Neuropsychopharmacology 2020, 45, 1963–1964. [Google Scholar] [CrossRef] [PubMed]
- Broadbear, J.H.; Sumpter, T.L.; Burke, T.F.; Husbands, S.M.; Lewis, J.W.; Woods, J.H.; Traynor, J.R. Methocinnamox is a potent, long-lasting, and selective antagonist of morphine-mediated antinociception in the mouse: Comparison with clocinnamox, beta-funaltrexamine, and beta-chlornaltrexamine. J. Pharmacol. Exp. Ther. 2000, 294, 933–940. [Google Scholar]
- Maguire, D.R.; Gerak, L.R.; Sanchez, J.J.; Javors, M.A.; Disney, A.; Husbands, S.M.; France, C.P. Effects of acute and repeated treatment with methocinnamox, a mu opioid receptor antagonist, on fentanyl self-administration in rhesus monkeys. Neuropsychopharmacology 2020, 45, 1986–1993. [Google Scholar] [CrossRef]
- Jimenez, V.M., Jr.; Castaneda, G.; France, C.P. Methocinnamox Reverses and Prevents Fentanyl-Induced Ventilatory Depression in Rats. J. Pharmacol. Exp. Ther. 2021, 377, 29–38. [Google Scholar] [CrossRef]
- Gerak, L.R.; Maguire, D.R.; Woods, J.H.; Husbands, S.M.; Disney, A.; France, C.P. Reversal and Prevention of the Respiratory-Depressant Effects of Heroin by the Novel mu-Opioid Receptor Antagonist Methocinnamox in Rhesus Monkeys. J. Pharmacol. Exp. Ther. 2019, 368, 229–236. [Google Scholar] [CrossRef]
- Maguire, D.R.; Gerak, L.R.; Woods, J.H.; Husbands, S.M.; Disney, A.; France, C.P. Long-Lasting Effects of Methocinnamox on Opioid Self-Administration in Rhesus Monkeys. J. Pharmacol. Exp. Ther. 2019, 368, 88–99. [Google Scholar] [CrossRef]
- Coe, M.A.; Pillitteri, J.L.; Sembower, M.A.; Gerlach, K.K.; Henningfield, J.E. Kratom as a substitute for opioids: Results from an online survey. Drug Alcohol. Depend. 2019, 202, 24–32. [Google Scholar] [CrossRef]
- Garcia-Romeu, A.; Cox, D.J.; Smith, K.E.; Dunn, K.E.; Griffiths, R.R. Kratom (Mitragyna speciosa): User demographics, use patterns, and implications for the opioid epidemic. Drug Alcohol. Depend. 2020, 208, 107849. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, O. Patterns of Kratom use and health impact in the US-Results from an online survey. Drug Alcohol. Depend. 2017, 176, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Veltri, C.; Grundmann, O. Current perspectives on the impact of Kratom use. Subst. Abus. Rehabil. 2019, 10, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Drug Enforcement Administration. Kratom (Mitragynine speciose korth) (Street Names: Thang, Kakuam, Thom, Ketum, Biak). Unites States Department of Justice, Drug Enforcement Administration; 2013; Drug Enforcement Administration. In Drugs of Abuse. A DEA Resource Guide; United States Department of Justice, Drug Enforcement Administration: Washington, DC, USA, 2017; Volume 94. [Google Scholar]
- Kruegel, A.C.; Grundmann, O. The medicinal chemistry and neuropharmacology of kratom: A preliminary discussion of a promising medicinal plant and analysis of its potential for abuse. Neuropharmacology 2018, 134, 108–120. [Google Scholar] [CrossRef]
- Takayama, H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem. Pharm. Bull. 2004, 52, 916–928. [Google Scholar] [CrossRef]
- Kruegel, A.C.; Gassaway, M.M.; Kapoor, A.; Varadi, A.; Majumdar, S.; Filizola, M.; Javitch, J.A.; Sames, D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. [Google Scholar] [CrossRef]
- Hemby, S.E.; McIntosh, S.; Leon, F.; Cutler, S.J.; McCurdy, C.R. Abuse liability and therapeutic potential of the Mitragyna speciosa (kratom) alkaloids mitragynine and 7-hydroxymitragynine. Addict. Biol. 2019, 24, 874–885. [Google Scholar] [CrossRef]
- Yue, K.; Kopajtic, T.A.; Katz, J.L. Abuse liability of mitragynine assessed with a self-administration procedure in rats. Psychopharmacology 2018, 235, 2823–2829. [Google Scholar] [CrossRef]
- Trakulsrichai, S.; Sathirakul, K.; Auparakkitanon, S.; Krongvorakul, J.; Sueajai, J.; Noumjad, N.; Sukasem, C.; Wananukul, W. Pharmacokinetics of mitragynine in man. Drug Des. Dev. Ther. 2015, 9, 2421–2429. [Google Scholar] [CrossRef]
- Henningfield, J.E.; Fant, R.V.; Wang, D.W. The abuse potential of kratom according the 8 factors of the controlled substances act: Implications for regulation and research. Psychopharmacology 2018, 235, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Daksla, N.; Wang, A.; Jin, Z.; Gupta, A.; Bergese, S.D. Oliceridine for the Management of Moderate to Severe Acute Postoperative Pain: A Narrative Review. Drug Des. Dev. Ther. 2023, 17, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Goudra, B. Oliceridine-Opioid of the 21(st) Century. Saudi. J. Anaesth. 2022, 16, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; van Dam, C.J.; Niesters, M.; van Velzen, M.; Fossler, M.J.; Demitrack, M.A.; Olofsen, E. Benefit and Risk Evaluation of Biased mu-Receptor Agonist Oliceridine versus Morphine. Anesthesiology 2020, 133, 559–568. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef]
- Miyano, K.; Manabe, S.; Komatsu, A.; Fujii, Y.; Mizobuchi, Y.; Uezono, E.; Ohshima, K.; Nonaka, M.; Kuroda, Y.; Narita, M.; et al. The G Protein Signal-Biased Compound TRV130; Structures, Its Site of Action and Clinical Studies. Curr. Top. Med. Chem. 2020, 20, 2822–2829. [Google Scholar] [CrossRef]
- Chen, X.T.; Pitis, P.; Liu, G.; Yuan, C.; Gotchev, D.; Cowan, C.L.; Rominger, D.H.; Koblish, M.; Dewire, S.M.; Crombie, A.L.; et al. Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl](2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl)amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 2013, 56, 8019–8031. [Google Scholar] [CrossRef]
- Oliceridine Briefing Document. FDA Advisory Committee Meeting. 11 October 2018. Available online: https://www.fda.gov/media/121230/download (accessed on 10 April 2023).
- Food and Drug Administration (FDA). Highlights of Prescribing Information—Olinvyk (Oliceridine). 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/210730s001lbl.pdf (accessed on 26 March 2023).
- Liang, D.Y.; Li, W.W.; Nwaneshiudu, C.; Irvine, K.A.; Clark, J.D. Pharmacological Characters of Oliceridine, a mu-Opioid Receptor G-Protein-Biased Ligand in Mice. Anesth. Analg. 2019, 129, 1414–1421. [Google Scholar] [CrossRef]
- Altarifi, A.A.; David, B.; Muchhala, K.H.; Blough, B.E.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef]
- Soergel, D.G.; Subach, R.A.; Sadler, B.; Connell, J.; Marion, A.S.; Cowan, C.L.; Violin, J.D.; Lark, M.W. First clinical experience with TRV130: Pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Simons, P.; van der Schrier, R.; van Lemmen, M.; Jansen, S.; Kuijpers, K.W.K.; van Velzen, M.; Sarton, E.; Nicklas, T.; Michalsky, C.; Demitrack, M.A.; et al. Respiratory Effects of Biased Ligand Oliceridine in Older Volunteers: A Pharmacokinetic-Pharmacodynamic Comparison with Morphine. Anesthesiology 2023, 138, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Nafziger, A.N.; Arscott, K.A.; Cochrane, K.; Skobieranda, F.; Burt, D.A.; Fossler, M.J. The Influence of Renal or Hepatic Impairment on the Pharmacokinetics, Safety, and Tolerability of Oliceridine. Clin. Pharmacol. Drug Dev. 2020, 9, 639–650. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Webster, L.; Kuss, M.; Daniels, S.; Bolognese, J.A.; Zuckerman, S.; Soergel, D.G.; Subach, R.A.; Cook, E.; Skobieranda, F. A randomized, phase 2 study investigating TRV130, a biased ligand of the mu-opioid receptor, for the intravenous treatment of acute pain. Pain 2016, 157, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.; Minkowitz, H.S.; Soergel, D.G.; Burt, D.A.; Subach, R.A.; Salamea, M.Y.; Fossler, M.J.; Skobieranda, F. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel micro-receptor G-protein pathway selective (mu-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res. 2017, 10, 2413–2424. [Google Scholar] [CrossRef]
- Viscusi, E.R.; Skobieranda, F.; Soergel, D.G.; Cook, E.; Burt, D.A.; Singla, N. APOLLO-1: A randomized placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein-biased ligand at the micro-opioid receptor, for management of moderate-to-severe acute pain following bunionectomy. J. Pain Res. 2019, 12, 927–943. [Google Scholar] [CrossRef] [PubMed]
- Singla, N.K.; Skobieranda, F.; Soergel, D.G.; Salamea, M.; Burt, D.A.; Demitrack, M.A.; Viscusi, E.R. APOLLO-2: A Randomized, Placebo and Active-Controlled Phase III Study Investigating Oliceridine (TRV130), a G Protein-Biased Ligand at the mu-Opioid Receptor, for Management of Moderate to Severe Acute Pain Following Abdominoplasty. Pain Pract. 2019, 19, 715–731. [Google Scholar] [CrossRef]
- Bergese, S.D.; Brzezinski, M.; Hammer, G.B.; Beard, T.L.; Pan, P.H.; Mace, S.E.; Berkowitz, R.D.; Cochrane, K.; Wase, L.; Minkowitz, H.S.; et al. ATHENA: A Phase 3, Open-Label Study Of The Safety And Effectiveness Of Oliceridine (TRV130), A G-Protein Selective Agonist At The micro-Opioid Receptor, In Patients With Moderate To Severe Acute Pain Requiring Parenteral Opioid Therapy. J. Pain Res. 2019, 12, 3113–3126. [Google Scholar] [CrossRef]
- Brzezinski, M.; Hammer, G.B.; Candiotti, K.A.; Bergese, S.D.; Pan, P.H.; Bourne, M.H.; Michalsky, C.; Wase, L.; Demitrack, M.A.; Habib, A.S. Low Incidence of Opioid-Induced Respiratory Depression Observed with Oliceridine Regardless of Age or Body Mass Index: Exploratory Analysis from a Phase 3 Open-Label Trial in Postsurgical Pain. Pain Ther. 2021, 10, 457–473. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; He, B.; He, X.; Zhou, X.E.; Guo, S.; Rao, Q.; Yang, J.; Liu, J.; Zhou, Q.; et al. Molecular recognition of morphine and fentanyl by the human mu-opioid receptor. Cell 2022, 185, 4361–4375. [Google Scholar] [CrossRef]
- Robertson, M.J.; Papasergi-Scott, M.M.; He, F.; Seven, A.B.; Meyerowitz, J.G.; Panova, O.; Peroto, M.C.; Che, T.; Skiniotis, G. Structure determination of inactive-state GPCRs with a universal nanobody. Nat. Struct. Mol. Biol. 2022, 29, 1188–1195. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Receptor | Main Functions | Tissue of Expression | Endogenous Ligand | Drugs a |
|---|---|---|---|---|
| μ-opioid receptor μ1; μ2; μ3 | pain supraspinal analgesia reward euphoria respiratory depression gastrointestinal motility anorexia pruritus urinary retention physical dependence | brain sub-mucosal plexus mesenteric plexus spinal cord | endomorphins endorphins enkephalins | morphine (agonist) methadone (agonist) fentanyl (agonist) heroin (agonist) buprenorphine (partial agonist) nalmefene (inverse agonist) cebranopadol (agonist) naloxone (antagonist) naltrexone (antagonist) |
| κ-opioid receptor κ1; κ2; κ3 | pain spinal analgesia sedation stress miosis dyspnea respiratory depression dysphoria anhedonia aversion dependence/addiction | brain mesenteric plexus peripheral sensory neurons | dynorphins | morphine (agonist) methadone (agonist) fentanyl (agonist) heroin (agonist) nalmefene (partial agonist) cebranopadol (partial agonist) naloxone (antagonist) buprenorphine (antagonist) naltrexone (antagonist) |
| δ-opioid receptor δ1; δ2 | pain analgesia antidepressant/anxiolytic effects | brain mesenteric plexus spinal cord peripheral sensory neurons | met-enkephalin leu-enkephalin endorphins | methadone (agonist) fentanyl (agonist) heroin (agonist) cebranopadol (agonist) naloxone (antagonist) buprenorphine (antagonist) naltrexone (antagonist) |
| opioid receptor-like 1 (ORL1) | pain, inflammatory and neuropathic pain fibromyalgia | forebrain (cortical areas, olfactory regions, limbic structures, thalamus) | nociception/orphanin FQ (N/OFQ) | cebranopadol (partial agonist) buprenorphine (partial agonist) |
| Study | Subjects | Dosing Regimen | Results | Adverse Events | Ref | |
|---|---|---|---|---|---|---|
| Phase I | First-in-human study | 74 healthy volunteers |
|
|
| [177] |
| Single-center, randomized, double-blind, placebo-controlled, 5-period, crossover study | 30 healthy men aged 18 to 50 years, with body mass indices of 19.0 to 32.0 kg/m2. |
|
|
| [131] | |
| Abuse liability study Single-dose, randomized, double-blind crossover trial | 60 healthy, non-dependent, recreational opioid users |
|
| |||
| Four-arm double-blind, randomized, crossover study | 18 healthy male and female volunteers aged 55 to 89 years |
|
|
| [178] | |
| Open-label, single-dose trial | 51 subjects, males or females, aged 18–80 years, BMI 18.0–35.0 kg/m2, a minimum weight of 50 kg; controls, age- and sex-matched at a ratio of 1:1 |
|
|
| [179] | |
| Phase II | Randomized, placebo- and active-controlled trial | 141 patients in the pilot phase, 192 patients in phase II, experiencing moderate-to-severe acute pain after bunionectomy |
|
|
| [180] |
| Randomized, double-blind, patient-controlled analgesia trial (NCT02335294) | 200 patients, males or females (99%) aged 18–65 years who planned to undergo abdominoplasty without any additional collateral procedures were enrolled |
|
|
| [181] | |
| Phase III | APOLLO-1: multicenter, double-blind, randomized, placebo- and active-controlled trial (NCT02815709) | 389 patients, males and females (84.8%), aged 18–75 years and scheduled to undergo primary, unilateral, first metatarsal bunionectomy |
|
|
| [182] |
| APOLLO-2: double-blind, randomized, placebo- and active-controlled trial (NCT02820324) | 401 patients, males and females (99.3%), aged 18–75 years, BMI < 35 kg/m2 or body weight > 40 kg, who followed abdominoplasty procedure with no additional collateral procedures |
|
|
| [183] | |
| ATHENA: multicenter, open-label trial (NCT02656875) | 768 patients, males and females (65%), >18 years, Caucasian (78%), surgical and non-surgical patients with painful medical conditions |
|
|
| [184] | |
| Phase IV | VOLITION: A multicenter pilot cohort study | 200 patients, males and females ≥18 years |
| In progress
| In progress | NCT04979247 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabanelli, R.; Brogi, S.; Calderone, V. Targeting Opioid Receptors in Addiction and Drug Withdrawal: Where Are We Going? Int. J. Mol. Sci. 2023, 24, 10888. https://doi.org/10.3390/ijms241310888
Tabanelli R, Brogi S, Calderone V. Targeting Opioid Receptors in Addiction and Drug Withdrawal: Where Are We Going? International Journal of Molecular Sciences. 2023; 24(13):10888. https://doi.org/10.3390/ijms241310888
Chicago/Turabian StyleTabanelli, Rita, Simone Brogi, and Vincenzo Calderone. 2023. "Targeting Opioid Receptors in Addiction and Drug Withdrawal: Where Are We Going?" International Journal of Molecular Sciences 24, no. 13: 10888. https://doi.org/10.3390/ijms241310888
APA StyleTabanelli, R., Brogi, S., & Calderone, V. (2023). Targeting Opioid Receptors in Addiction and Drug Withdrawal: Where Are We Going? International Journal of Molecular Sciences, 24(13), 10888. https://doi.org/10.3390/ijms241310888