
Skin Wound following Irradiation Aggravates Radiation-Induced Brain Injury in a Mouse Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
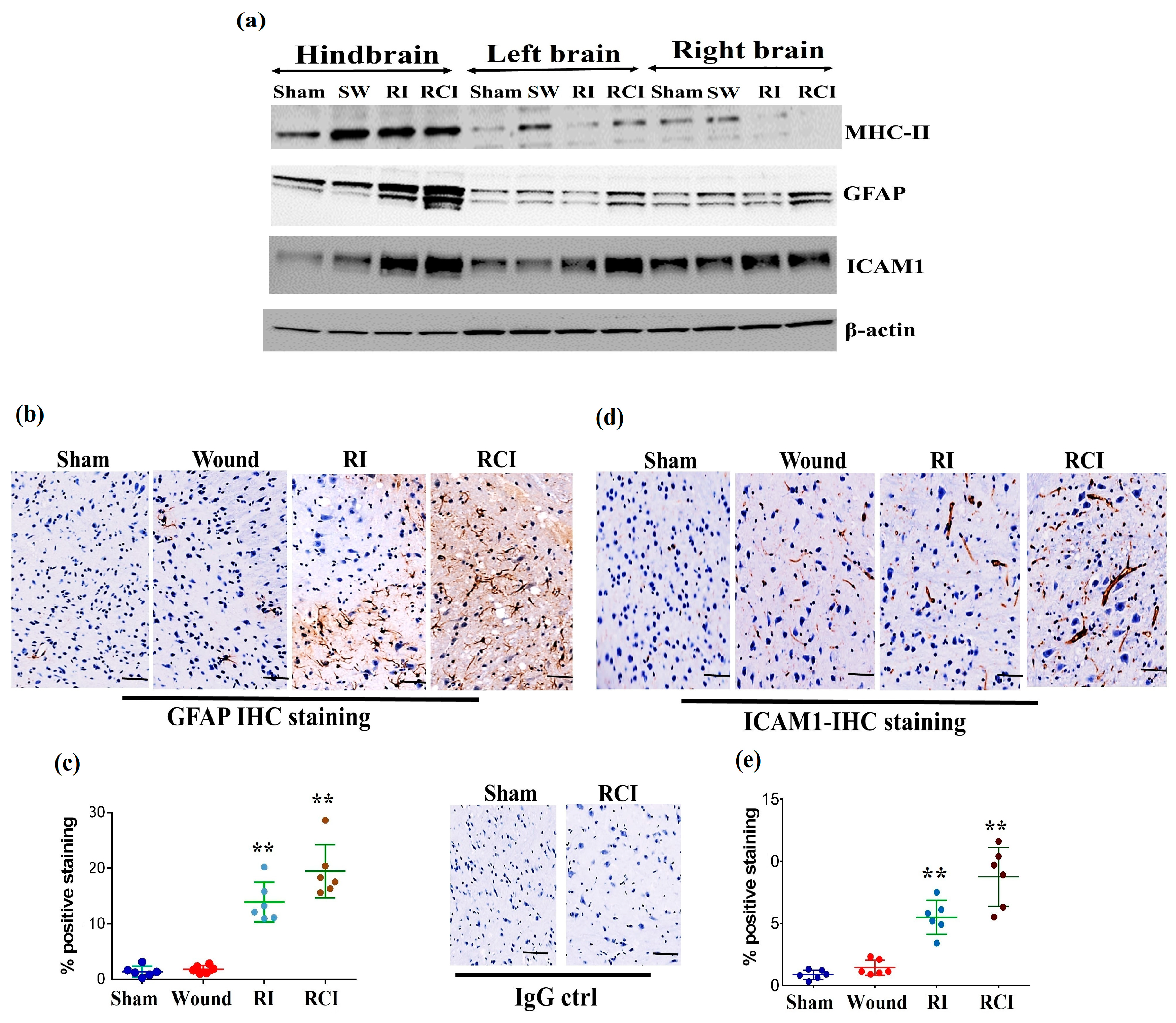{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Survival, Body Weight, and/or Skin Wound Healing after SW, RI, and RCI
2.2. RI- and RCI-Induced Proinflammatory Chemokines Increase in Mouse Brain
2.3. RI and RCI Upregulate Proinflammatory Factor IP-10 and Cyclin-Dependent Kinase Inhibitor p21 in Mouse Brain
2.4. RI and RCI Induce Mouse Blood–Brain Barrier (BBB) Leakage
2.5. RCI Induced BBB Leakage, as Shown by GFAP Increase in Mouse Serum
2.6. RCI Causes More Severe Neural Cell Loss Than RI
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Mice and Animal Care
4.3. Total-Body Irradiation
4.4. Skin Wounding
4.5. Survival, Body Weight, and Wound Healing
4.6. Brain Tissue Collection
4.7. Brain Cytokine Antibody Array
4.8. Protein Extraction and Immunoblotting
4.9. Brain Immunohistochemistry (IHC) Staining
4.10. Enzyme-Linked Immune Sorbent Assay (ELISA)
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DiCarlo, A.L.; Hatchett, R.J.; Kaminski, J.M.; Ledney, G.D.; Pellmar, T.C.; Okunieff, P.; Ramakrishnan, N. Medical Countermeasures for Radiation Combined Injury: Radiation with Burn, Blast, Trauma and/or Sepsis. Report of an NIAID Workshop, March 26–27, 2007. Radiat. Res. 2008, 169, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhai, M.; Lin, B.; Cui, W.; Hull, L.; Li, X.; Anderson, M.N.; Smith, J.T.; Umali, M.V.; Jiang, S.; et al. PEG-G-CSF and L-Citrulline Combination Therapy for Mitigating Skin Wound Combined Radiation Injury in a Mouse Model. Radiat. Res. 2021, 196, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, B.; Zhai, M.; Cui, W.; Hull, L.; Zizzo, A.; Li, X.; Kiang, J.G.; Xiao, M. Deteriorative Effects of Radiation Injury Combined with Skin Wounding in a Mouse Model. Toxics 2022, 10, 785. [Google Scholar] [CrossRef] [PubMed]
- Hauer-Jensen, M.; Kumar, K.S.; Wang, J.; Berbee, M.; Fu, Q.; Boerma, M. Intestinal Toxicity in Radiation- and Combined Injury: Significance, Mechanisms, and Countermeasures: 2008; Nova Science Publishers, Inc.: New York, NY, USA, 2008; pp. 61–100. [Google Scholar]
- Kiang, J.G.; Jiao, W.; Cary, L.H.; Mog, S.R.; Elliott, T.B.; Pellmar, T.C.; Ledney, G.D. Wound Trauma Increases Radiation-Induced Mortality by Activation of iNOS Pathway and Elevation of Cytokine Concentrations and Bacterial Infection. Radiat. Res. 2010, 173, 319–332. [Google Scholar] [CrossRef]
- Pellmar, T.C.; Rockwell, S. Priority List of Research Areas for Radiological Nuclear Threat Countermeasures. Radiat. Res. 2005, 163, 115–123. [Google Scholar] [CrossRef]
- Li, X.; Cui, W.; Hull, L.; Smith, J.T.; Kiang, J.G.; Xiao, M. Effects of Low-to-Moderate Doses of Gamma Radiation on Mouse Hematopoietic System. Radiat. Res. 2018, 190, 612–622. [Google Scholar] [CrossRef]
- Ha, C.T.; Li, X.-H.; Fu, D.; Moroni, M.; Fisher, C.; Arnott, R.; Srinivasan, V.; Xiao, M. Circulating Interleukin-18 as a Biomarker of Total-Body Radiation Exposure in Mice, Minipigs, and Nonhuman Primates (NHP). PLoS ONE 2014, 9, e109249. [Google Scholar] [CrossRef]
- Cui, W.; Hull, L.; Zizzo, A.; Wang, L.; Lin, B.; Zhai, M.; Xiao, M. Pharmacokinetic Study of rhIL-18BP and Its Effect on Radiation-Induced Cytokine Changes in Mouse Serum and Intestine. Toxics 2022, 11, 35. [Google Scholar] [CrossRef]
- Lénárt, N.; Brough, D.; Dénes, Á. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J. Cereb. Blood Flow Metab. 2016, 36, 1668–1685. [Google Scholar] [CrossRef]
- Gorbunov, N.V.; Kiang, J.G. Brain Damage and Patterns of Neurovascular Disorder after Ionizing Irradiation. Complications in Radiotherapy and Radiation Combined Injury. Radiat. Res. 2021, 196, 1–16. [Google Scholar] [CrossRef]
- Balentova, S.; Adamkov, M. Molecular, Cellular and Functional Effects of Radiation-Induced Brain Injury: A Review. Int. J. Mol. Sci. 2015, 16, 27796–27815. [Google Scholar] [CrossRef]
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neuro-Oncol. Adv. 2020, 2, vdaa057. [Google Scholar] [CrossRef]
- Boyd, A.; Byrne, S.; Middleton, R.J.; Banati, R.B.; Liu, G.-J. Control of Neuroinflammation through Radiation-Induced Microglial Changes. Cells 2021, 10, 2381. [Google Scholar] [CrossRef]
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288. [Google Scholar] [CrossRef]
- Seth, P.; Koul, N. Astrocyte, the star avatar: Redefined. J. Biosci. 2008, 33, 405–421. [Google Scholar] [CrossRef]
- Pullagurla, S.R.; Baird, A.E.; Adamski, M.G.; A Soper, S. Current and future bioanalytical approaches for stroke assessment. Bioanalysis 2015, 7, 1017–1035. [Google Scholar] [CrossRef]
- Lupo, J.M.; Molinaro, A.M.; Essock-Burns, E.; Butowski, N.; Chang, S.M.; Cha, S.; Nelson, S.J. The effects of anti-angiogenic therapy on the formation of radiation-induced microbleeds in normal brain tissue of patients with glioma. Neuro-Oncology 2015, 18, 87–95. [Google Scholar] [CrossRef]
- Kiang, J.G.; Smith, J.T.; Anderson, M.N.; Umali, M.V.; Ho, C.; Zhai, M.; Lin, B.; Jiang, S. A novel therapy, using Ghrelin with pegylated G-CSF, inhibits brain hemorrhage from ionizing radiation or combined radiation injury. Pharm. Pharmacol. Int. J. 2019, 7, 62–65. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef]
- Lumniczky, K.; Szatmári, T.; Sáfrány, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. [Google Scholar] [CrossRef]
- Tofilon, P.J.; Fike, J.R. The Radioresponse of the Central Nervous System: A Dynamic Process. Radiat. Res. 2000, 153, 357–370. [Google Scholar] [CrossRef]
- Greene-Schloesser, D.; Robbins, M.E.; Peiffer, A.M.; Shaw, E.G.; Wheeler, K.T.; Chan, M.D. Radiation-induced brain injury: A review. Front. Oncol. 2012, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Sándor, N.; Walter, F.R.; Bocsik, A.; Sántha, P.; Schilling-Tóth, B.; Léner, V.; Varga, Z.; Kahán, Z.; Deli, M.; Sáfrány, G.; et al. Low Dose Cranial Irradiation-Induced Cerebrovascular Damage Is Reversible in Mice. PLoS ONE 2014, 9, e112397. [Google Scholar] [CrossRef] [PubMed]
- Markarian, M.; Krattli, R.P.; Baddour, J.D.; Alikhani, L.; Giedzinski, E.; Usmani, M.T.; Agrawal, A.; Baulch, J.E.; Tenner, A.J.; Acharya, M.M. Glia-Selective Deletion of Complement C1q Prevents Radiation-Induced Cognitive Deficits and Neuroinflammation. Cancer Res. 2020, 81, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.-H.; Kim, J.-H.; Seo, J.-W.; Han, H.-S.; Lee, W.-H.; Mori, K.; Nakao, K.; Barasch, J.; Suk, K. Lipocalin-2 Is a Chemokine Inducer in the Central Nervous System: ROLE OF CHEMOKINE LIGAND 10 (CXCL10) IN LIPOCALIN-2-INDUCED CELL MIGRATION*. J. Biol. Chem. 2011, 286, 43855–43870. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Cheng, B.Z.; Zheng, Y.Z.; Li, M.Y.-Q.; Wong, C.S. Cellular Senescence in Mouse Hippocampus After Irradiation and the Role of p53 and p21. J. Neuropathol. Exp. Neurol. 2017, 76, 260–269. [Google Scholar] [CrossRef]
- Kiang, J.G.; Blakely, W.F. Combined radiation injury and its impacts on radiation countermeasures and biodosimetry. Int. J. Radiat. Biol. 2023, 1–11. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 1–20. [Google Scholar] [CrossRef]
- Ambrosini, E.; Aloisi, F. Chemokines and Glial Cells: A Complex Network in the Central Nervous System. Neurochem. Res. 2004, 29, 1017–1038. [Google Scholar] [CrossRef]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-Inducible Protein 10 (IP-10; CXCL10)-Deficient Mice Reveal a Role for IP-10 in Effector T Cell Generation and Trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef]
- Medoff, B.D.; Sauty, A.; Tager, A.M.; Maclean, J.A.; Smith, R.N.; Mathew, A.; Dufour, J.H.; Luster, A.D. IFN-γ-Inducible Protein 10 (CXCL10) Contributes to Airway Hyperreactivity and Airway Inflammation in a Mouse Model of Asthma. J. Immunol. 2002, 168, 5278–5286. [Google Scholar] [CrossRef]
- Biber, K.; Dijkstra, I.; Trebst, C.; De Groot, C.; Ransohoff, R.; Boddeke, H. Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience 2002, 112, 487–497. [Google Scholar] [CrossRef]
- Berthoud, T.K.; Dunachie, S.J.; Todryk, S.; Hill, A.V.; Fletcher, H.A. MIG (CXCL9) is a more sensitive measure than IFN-γ of vaccine induced T-cell responses in volunteers receiving investigated malaria vaccines. J. Immunol. Methods 2009, 340, 33–41. [Google Scholar] [CrossRef]
- Li, X.; Cui, W.; Hull, L.; Wang, L.; Yu, T.; Xiao, M. IL-18 binding protein (IL-18BP) as a novel radiation countermeasure after radiation exposure in mice. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Ivanovska, M.; Abdi, Z.; Murdjeva, M.; Macedo, D.; Maes, A.; Maes, M. CCL-11 or Eotaxin-1: An Immune Marker for Ageing and Accelerated Ageing in Neuro-Psychiatric Disorders. Pharmaceuticals 2020, 13, 230. [Google Scholar] [CrossRef]
- A Hedrick, J.A.; Zlotnik, A. Identification and characterization of a novel beta chemokine containing six conserved cysteines. J. Immunol. 1997, 159, 1589–1593. [Google Scholar] [CrossRef]
- Mojsilovic-Petrovic, J.; Callaghan, D.; Cui, H.; Dean, C.; Stanimirovic, D.B.; Zhang, W. Hypoxia-inducible factor-1 (HIF-1) is involved in the regulation of hypoxia-stimulated expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in astrocytes. J. Neuroinflammation 2007, 4, 12. [Google Scholar] [CrossRef]
- Bednarek, N.; Svedin, P.; Garnotel, R.; Favrais, G.; Loron, G.; Schwendiman, L.; Hagberg, H.; Morville, P.; Mallard, C.; Gressens, P. Increased MMP-9 and TIMP-1 in mouse neonatal brain and plasma and in human neonatal plasma after hypoxia–ischemia: A potential marker of neonatal encephalopathy. Pediatr. Res. 2011, 71, 63–70. [Google Scholar] [CrossRef]
- Cartier, L.; Hartley, O.; Dubois-Dauphin, M.; Krause, K.-H. Chemokine receptors in the central nervous system: Role in brain inflammation and neurodegenerative diseases. Brain Res. Rev. 2005, 48, 16–42. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M. Role of GFAP in CNS injuries. Neurosci. Lett. 2014, 565, 7–13. [Google Scholar] [CrossRef] [PubMed]
- McMahon, P.J.; Panczykowski, D.M.; Yue, J.K.; Puccio, A.M.; Inoue, T.; Sorani, M.D.; Lingsma, H.F.; Maas, A.I.R.; Valadka, A.B.; Yuh, E.L.; et al. Measurement of the Glial Fibrillary Acidic Protein and Its Breakdown Products GFAP-BDP Biomarker for the Detection of Traumatic Brain Injury Compared to Computed Tomography and Magnetic Resonance Imaging. J. Neurotrauma 2015, 32, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Anbarasan, C.; Bavanilatha, M.; Latchumanadhas, K.; Mullasari, S.A. ICAM-1 molecular mechanism and genome wide SNP’s association studies. Indian Hear. J. 2015, 67, 282–287. [Google Scholar] [CrossRef]
- Regal-McDonald, K.; Somarathna, M.; Lee, T.; Litovsky, S.H.; Barnes, J.; Peretik, J.M.; Traylor, J.G.; Orr, A.; Patel, R.P. Assessment of ICAM-1 N-glycoforms in mouse and human models of endothelial dysfunction. PLoS ONE 2020, 15, e0230358. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Ayanlaja, A.A.; Xiong, Y.; Gao, Y.; Ji, G.; Tang, C.; Abdullah, Z.A.; Gao, D. Distinct Features of Doublecortin as a Marker of Neuronal Migration and Its Implications in Cancer Cell Mobility. Front. Mol. Neurosci. 2017, 10, 199. [Google Scholar] [CrossRef]
- Li, X.H.; Ha, C.T.; Fu, D.; Landauer, M.R.; Ghosh, S.P.; Xiao, M. Delta-Tocotrienol Suppresses Radiation-Induced MicroRNA-30 and Protects Mice and Human CD34+ Cells from Radiation Injury. PLoS ONE 2015, 10, e0122258. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Li, X.; Wang, L.; Lin, B.; Zhai, M.; Hull, L.; Zizzo, A.; Cui, W.; Kiang, J.G. Skin Wound following Irradiation Aggravates Radiation-Induced Brain Injury in a Mouse Model. Int. J. Mol. Sci. 2023, 24, 10701. https://doi.org/10.3390/ijms241310701
Xiao M, Li X, Wang L, Lin B, Zhai M, Hull L, Zizzo A, Cui W, Kiang JG. Skin Wound following Irradiation Aggravates Radiation-Induced Brain Injury in a Mouse Model. International Journal of Molecular Sciences. 2023; 24(13):10701. https://doi.org/10.3390/ijms241310701
Chicago/Turabian StyleXiao, Mang, Xianghong Li, Li Wang, Bin Lin, Min Zhai, Lisa Hull, Alex Zizzo, Wanchang Cui, and Juliann G. Kiang. 2023. "Skin Wound following Irradiation Aggravates Radiation-Induced Brain Injury in a Mouse Model" International Journal of Molecular Sciences 24, no. 13: 10701. https://doi.org/10.3390/ijms241310701
APA StyleXiao, M., Li, X., Wang, L., Lin, B., Zhai, M., Hull, L., Zizzo, A., Cui, W., & Kiang, J. G. (2023). Skin Wound following Irradiation Aggravates Radiation-Induced Brain Injury in a Mouse Model. International Journal of Molecular Sciences, 24(13), 10701. https://doi.org/10.3390/ijms241310701