
Expression-Based Diagnosis, Treatment Selection, and Drug Development for Breast Cancer

 ,
,
Abstract
1. Introduction
2. Results
2.1. 28-Gene Signature as a Diagnostic Assay for Breast Cancer
2.2. RT-PCR Validation of 26 Genes out of the 28-Gene Signature in Breast Cancer Tissue Samples
2.3. RT-PCR Validation of 28-Gene Signature in Breast Cancer Blood Samples
2.4. Prognosis within Breast Cancer Subtypes Using Next-Generation Sequencing Data
2.5. Correlation of Signature Genes with Immune Infiltration
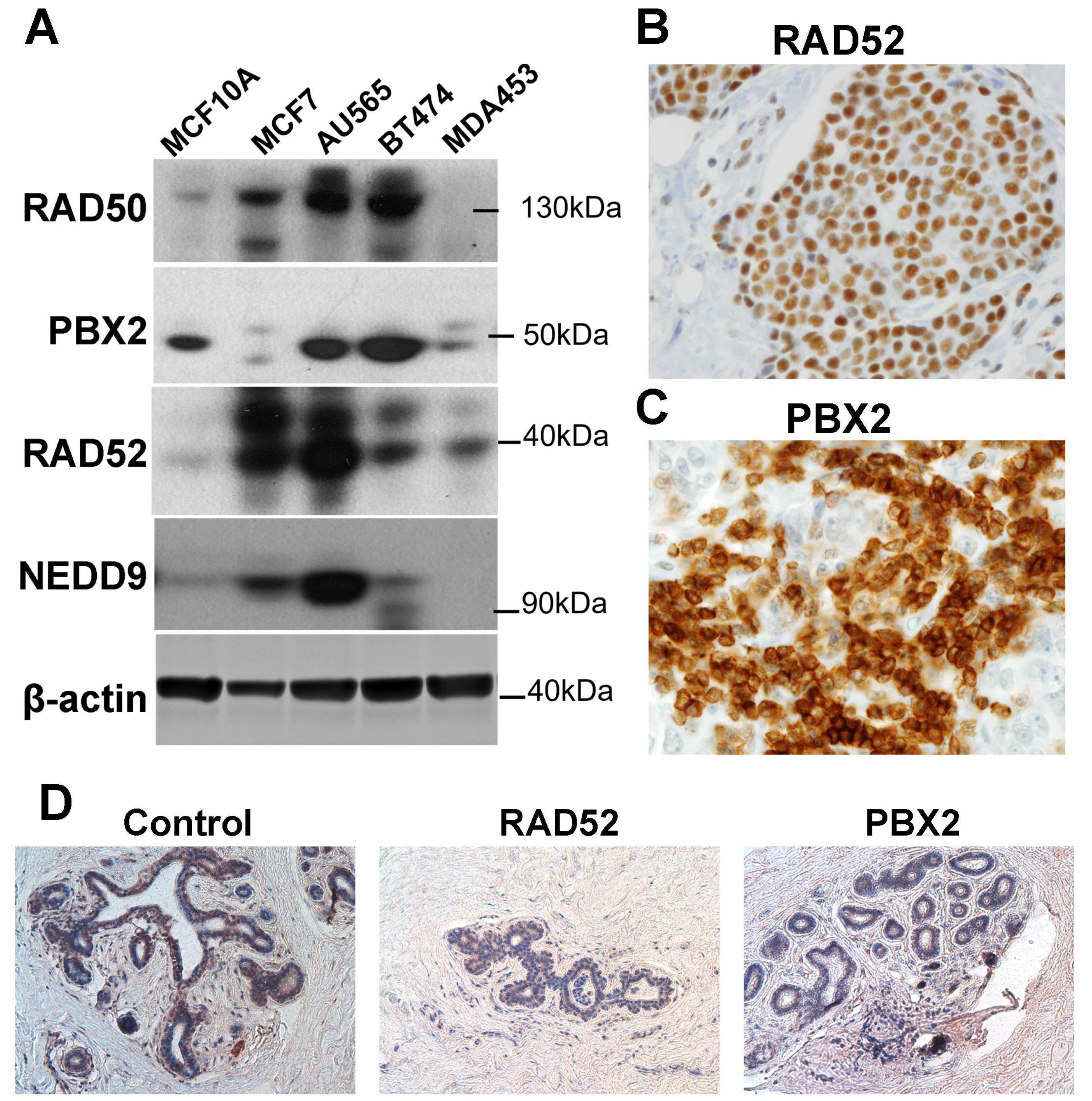
2.6. Protein Expression Validation of the Signature Genes
2.7. Proliferation Potential of the Signature Genes
2.8. Association with Drug Response
2.9. Discovery of New Drugs with CMap
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Blood Collection
4.3. RNA Extraction, Quality, and Concentration Assessment
4.4. Generation of Complementary DNA (cDNA)
4.5. Real-Time RT-PCR Low-Density Arrays
4.6. Statistical Analysis of Real-Time RT-PCR Data
4.7. Stromal and Immune Infiltration
4.8. Protein Expression Validation Using Western Blot Analysis
4.9. Protein Expression Validation Using Immunohistochemistry Analysis
4.10. Microarray Data Processing
4.11. Construction of Molecular Classifiers
4.12. Time-Dependent Receiver Operating Characteristics (ROC) Curves and Area under the ROC Curve (AUC)
4.13. Clustering Analysis
4.14. Proliferation Assays
4.15. Cancer Cell Line Encyclopedia (CCLE)
4.16. Drug Sensitivity in CCLE
4.17. CMap
4.18. Statistics Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- van de Vijver, M.J.; He, Y.D.; van ‘t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- van ‘t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Sparano, J.A.; Gray, R.J.; Ravdin, P.M.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; et al. Clinical and Genomic Risk to Guide the Use of Adjuvant Therapy for Breast Cancer. N. Engl. J. Med. 2019, 380, 2395–2405. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; Olson, J.A., Jr.; et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef]
- Perou, C.M.; Jeffrey, S.S.; van de Rijn, M.; Rees, C.A.; Eisen, M.B.; Ross, D.T.; Pergamenschikov, A.; Williams, C.F.; Zhu, S.X.; Lee, J.C.; et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc. Natl. Acad. Sci. USA 1999, 96, 9212–9217. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Winslow, S.; Leandersson, K.; Edsjö, A.; Larsson, C. Prognostic stromal gene signatures in breast cancer. Breast Cancer Res. 2015, 17, 23. [Google Scholar] [CrossRef]
- Sgroi, D.C.; Sestak, I.; Cuzick, J.; Zhang, Y.; Schnabel, C.A.; Schroeder, B.; Erlander, M.G.; Dunbier, A.; Sidhu, K.; Lopez-Knowles, E.; et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: A prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol. 2013, 14, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef]
- Müller, B.M.; Keil, E.; Lehmann, A.; Winzer, K.J.; Richter-Ehrenstein, C.; Prinzler, J.; Bangemann, N.; Reles, A.; Stadie, S.; Schoenegg, W.; et al. The EndoPredict Gene-Expression Assay in Clinical Practice—Performance and Impact on Clinical Decisions. PLoS ONE 2013, 8, e68252. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Nuyten, D.S.; Sneddon, J.B.; Hastie, T.; Tibshirani, R.; Sorlie, T.; Dai, H.; He, Y.D.; van ‘t Veer, L.J.; Bartelink, H.; et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc. Natl. Acad. Sci. USA 2005, 102, 3738–3743. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007, 8, R157. [Google Scholar] [CrossRef]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. S2), S26–S35. [Google Scholar] [CrossRef]
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52 Pt 2, 178–188. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Choi, L. Breast Cancer. September 2022. Available online: https://www.merckmanuals.com/home/women-s-health-issues/breast-disorders/breast-cancer (accessed on 17 April 2023).
- Gøtzsche, P.C.; Jørgensen, K.J. Screening for breast cancer with mammography. Cochrane Database Syst. Rev. 2013, 2013, Cd001877. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.D.; Tyne, K.; Naik, A.; Bougatsos, C.; Chan, B.; Nygren, P.; Humphrey, L.U.S. Preventive Services Task Force Evidence Syntheses, formerly Systematic Evidence Reviews. In Screening for Breast Cancer: Systematic Evidence Review Update for the US Preventive Services Task Force; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2009. [Google Scholar]
- Shaevitch, D.; Taghipour, S.; Miller, A.B.; Montgomery, N.; Harvey, B. Tumor size distribution of invasive breast cancers and the sensitivity of screening methods in the Canadian National Breast Screening Study. J. Cancer Res. Ther. 2017, 13, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Laia, B.V.; Marcos, M.B.; Refael, C.M.; Francisco, S.C.; Jose, T.; Blai, B.S. Molecular Diagnosis of Sentinel Lymph Nodes for Breast Cancer: One Step Ahead for Standardization. Diagn. Mol. Pathol. 2011, 20, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Penault-Llorca, F.; Viale, G. Pathological and molecular diagnosis of triple-negative breast cancer: A clinical perspective. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23 (Suppl. S6), vi19–vi22. [Google Scholar] [CrossRef]
- Milanezi, F.; Carvalho, S.; Schmitt, F.C. EGFR/HER2 in breast cancer: A biological approach for molecular diagnosis and therapy. Expert Rev. Mol. Diagn. 2008, 8, 417–434. [Google Scholar] [CrossRef]
- Rakha, E.A.; Green, A.R. Molecular classification of breast cancer: What the pathologist needs to know. Pathology 2017, 49, 111–119. [Google Scholar] [CrossRef]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Fusco, N.; Geyer, F.C.; De Filippo, M.R.; Martelotto, L.G.; Ng, C.K.; Piscuoglio, S.; Guerini-Rocco, E.; Schultheis, A.M.; Fuhrmann, L.; Wang, L.; et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod. Pathol. 2016, 29, 1292–1305. [Google Scholar] [CrossRef]
- Gage, M.; Wattendorf, D.; Henry, L.R. Translational advances regarding hereditary breast cancer syndromes. J. Surg. Oncol. 2012, 105, 444–451. [Google Scholar] [CrossRef]
- Levenson, V.V. Biomarkers for early detection of breast cancer: What, when, and where? Biochim. Biophys. Acta 2007, 1770, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Qian, Y.; Wei, L.; Abraham, J.; Shi, X.; Castranova, V.; Harner, E.J.; Flynn, D.C.; Guo, L. Population-Based Molecular Prognosis of Breast Cancer by Transcriptional Profiling. Clin. Cancer Res. 2007, 13, 2014–2022. [Google Scholar] [CrossRef]
- Rathnagiriswaran, S.; Wan, Y.W.; Abraham, J.; Castranova, V.; Qian, Y.; Guo, N.L. A population-based gene signature is predictive of breast cancer survival and chemoresponse. Int. J. Oncol. 2010, 36, 607–616. [Google Scholar] [PubMed]
- Wan, Y.W.; Qian, Y.; Rathnagiriswaran, S.; Castranova, V.; Guo, N.L. A breast cancer prognostic signature predicts clinical outcomes in multiple tumor types. Oncol. Rep. 2010, 24, 489–494. [Google Scholar] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e1417. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Chen, D.T.; Nasir, A.; Culhane, A.; Venkataramu, C.; Fulp, W.; Rubio, R.; Wang, T.; Agrawal, D.; McCarthy, S.M.; Gruidl, M.; et al. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue 1. Breast Cancer Res. Treat. 2010, 119, 335–346. [Google Scholar] [CrossRef]
- Poola, I.; DeWitty, R.L.; Marshalleck, J.J.; Bhatnagar, R.; Abraham, J.; Leffall, L.D. Identification of MMP-1 as a putative breast cancer predictive marker by global gene expression analysis 1. Nat. Med. 2005, 11, 481–483. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Ye, Q.; Putila, J.; Raese, R.; Dong, C.; Qian, Y.; Dowlati, A.; Guo, N.L. Identification of Prognostic and Chemopredictive microRNAs for Non-Small-Cell Lung Cancer by Integrating SEER-Medicare Data. Int. J. Mol. Sci. 2021, 22, 7658. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Mellor, P.; Ward, A.K.; Kendall, S.; McDonald, M.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Napper, S.; Anderson, D.H. Molecular characterization of breast cancer cell lines through multiple omic approaches. Breast Cancer Res. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. MMBR 2002, 66, 630–670. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Golemis, E.A. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat. Cell Biol. 2005, 7, 937–946. [Google Scholar] [CrossRef]
- Izumchenko, E.; Singh, M.K.; Plotnikova, O.V.; Tikhmyanova, N.; Little, J.L.; Serebriiskii, I.G.; Seo, S.; Kurokawa, M.; Egleston, B.L.; Klein-Szanto, A.; et al. NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res. 2009, 69, 7198–7206. [Google Scholar] [CrossRef]
- Li, Y.; Bavarva, J.H.; Wang, Z.; Guo, J.; Qian, C.; Thibodeau, S.N.; Golemis, E.A.; Liu, W. HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene 2011, 30, 2633–2643. [Google Scholar] [CrossRef]
- Ghosh, S.; Hönscheid, A.; Dückers, G.; Ginzel, S.; Gohlke, H.; Gombert, M.; Kempkes, B.; Klapper, W.; Kuhlen, M.; Laws, H.J.; et al. Human RAD52—A novel player in DNA repair in cancer and immunodeficiency. Haematologica 2017, 102, e69–e72. [Google Scholar] [CrossRef]
- Monica, K.; Galili, N.; Nourse, J.; Saltman, D.; Cleary, M.L. PBX2 and PBX3, new homeobox genes with extensive homology to the human proto-oncogene PBX1. Mol. Cell. Biol. 1991, 11, 6149–6157. [Google Scholar] [CrossRef]
- Wright, E.K., Jr.; Page, S.H.; Barber, S.A.; Clements, J.E. Prep1/Pbx2 complexes regulate CCL2 expression through the -2578 guanine polymorphism. Genes Immun. 2008, 9, 419–430. [Google Scholar] [CrossRef]
- McDonald, J.A.; Goyal, A.; Terry, M.B. Alcohol Intake and Breast Cancer Risk: Weighing the Overall Evidence. Curr. Breast Cancer Rep. 2013, 5, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Miller, A.B.; Collishaw, N.E.; Palmer, J.R.; Hammond, S.K.; Salmon, A.G.; Cantor, K.P.; Miller, M.D.; Boyd, N.F.; Millar, J.; et al. Active smoking and secondhand smoke increase breast cancer risk: The report of the Canadian Expert Panel on Tobacco Smoke and Breast Cancer Risk (2009). Tob. Control 2011, 20, e2. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, V.S.; Hartmann, L.C.; Degnim, A.C.; Vierkant, R.A.; Ghosh, K.; Vachon, C.M.; Frost, M.H.; Maloney, S.D.; Reynolds, C.; Boughey, J.C. Assessment of the accuracy of the Gail model in women with atypical hyperplasia. J. Clin. Oncol. 2008, 26, 5374–5379. [Google Scholar] [CrossRef]
- Sa-Nguanraksa, D.; Sasanakietkul, T.; Chayanuch, O.; Kulprom, A.; Pornchai, O. Gail Model Underestimates Breast Cancer Risk in Thai Population. Asian Pac. J. Cancer Prev. 2019, 20, 2385–2389. [Google Scholar] [CrossRef]
- Li, K.; Anderson, G.; Viallon, V.; Arveux, P.; Kvaskoff, M.; Fournier, A.; Krogh, V.; Tumino, R.; Sánchez, M.J.; Ardanaz, E.; et al. Risk prediction for estrogen receptor-specific breast cancers in two large prospective cohorts. Breast Cancer Res. 2018, 20, 147. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hagrass, H.A.; Sharaf, S.; Pasha, H.F.; Tantawy, E.A.; Mohamed, R.H.; Kassem, R. Circulating microRNAs—A new horizon in molecular diagnosis of breast cancer. Genes Cancer 2015, 6, 281–287. [Google Scholar] [CrossRef]
- Huth, H.W.; Albarnaz, J.D.; Torres, A.A.; Bonjardim, C.A.; Ropert, C. MEK2 controls the activation of MKK3/MKK6-p38 axis involved in the MDA-MB-231 breast cancer cell survival: Correlation with cyclin D1 expression. Cell. Signal. 2016, 28, 1283–1291. [Google Scholar] [CrossRef]
- Van Swearingen, A.E.D.; Sambade, M.J.; Siegel, M.B.; Sud, S.; McNeill, R.S.; Bevill, S.M.; Chen, X.; Bash, R.E.; Mounsey, L.; Golitz, B.T.; et al. Combined kinase inhibitors of MEK1/2 and either PI3K or PDGFR are efficacious in intracranial triple-negative breast cancer. Neuro Oncol. 2017, 19, 1481–1493. [Google Scholar] [CrossRef]
- Kikuchi, K.; McNamara, K.M.; Miki, Y.; Iwabuchi, E.; Kanai, A.; Miyashita, M.; Ishida, T.; Sasano, H. S100P and Ezrin promote trans-endothelial migration of triple negative breast cancer cells. Cell. Oncol. 2019, 42, 67–80. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Z.; Liu, W.; Lei, R.; Shan, J.; Li, L.; Wang, X. Distinct prognostic values of S100 mRNA expression in breast cancer. Sci. Rep. 2017, 7, 39786. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Lyu, Y.; Jiang, J.; Wang, M.; Zhang, R.; Liew, C.C.; Wang, B.; Cheng, C. Peripheral blood transcriptome identifies high-risk benign and malignant breast lesions. PLoS ONE 2020, 15, e0233713. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, M.A.; Peltenburg, L.T.; Murre, C. Hox gene products modulate the DNA binding activity of Pbx1 and Pbx2. Mech. Dev. 1995, 52, 99–108. [Google Scholar] [CrossRef]
- Neuteboom, S.T.; Peltenburg, L.T.; van Dijk, M.A.; Murre, C. The hexapeptide LFPWMR in Hoxb-8 is required for cooperative DNA binding with Pbx1 and Pbx2 proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 9166–9170. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.C.; Errico, M.C.; Bottero, L.; Penkov, D.; Resnati, M.; Blasi, F.; Caré, A. Oncogenic HoxB7 requires TALE cofactors and is inactivated by a dominant-negative Pbx1 mutant in a cell-specific manner. Cancer Lett. 2008, 266, 144–155. [Google Scholar] [CrossRef]
- Giulianelli, S.; Riggio, M.; Guillardoy, T.; Pérez Piñero, C.; Gorostiaga, M.A.; Sequeira, G.; Pataccini, G.; Abascal, M.F.; Toledo, M.F.; Jacobsen, B.M.; et al. FGF2 induces breast cancer growth through ligand-independent activation and recruitment of ERα and PRBΔ4 isoform to MYC regulatory sequences. Int. J. Cancer 2019, 145, 1874–1888. [Google Scholar] [CrossRef]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef]
- Yiangou, C.; Gomm, J.J.; Coope, R.C.; Law, M.; Luqmani, Y.A.; Shousha, S.; Coombes, R.C.; Johnston, C.L. Fibroblast growth factor 2 in breast cancer: Occurrence and prognostic significance. Br. J. Cancer 1997, 75, 28–33. [Google Scholar] [CrossRef][Green Version]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef]
- Keller, G.; Schafhausen, P.; Brümmendorf, T.H. Bosutinib. Recent Results Cancer Res. 2010, 184, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Singh, N.; Mishra, N.; Nisha, R.; Alka; Maurya, P.; Pal, R.R.; Singh, S.; Saraf, S.A. Functionalized bosutinib liposomes for target specific delivery in management of estrogen-positive cancer. Colloids Surf. B Biointerfaces 2022, 218, 112763. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent Results Cancer Res. 2018, 212, 29–68. [Google Scholar] [CrossRef] [PubMed]
- Morris, P.G.; Rota, S.; Cadoo, K.; Zamora, S.; Patil, S.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; et al. Phase II Study of Paclitaxel and Dasatinib in Metastatic Breast Cancer. Clin. Breast Cancer 2018, 18, 387–394. [Google Scholar] [CrossRef]
- Gaule, P.; Mukherjee, N.; Corkery, B.; Eustace, A.J.; Gately, K.; Roche, S.; O’Connor, R.; O’Byrne, K.J.; Walsh, N.; Duffy, M.J.; et al. Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers 2019, 11, 548. [Google Scholar] [CrossRef]
- Xu, H.; Washington, S.; Verderame, M.F.; Manni, A. Role of non-receptor and receptor tyrosine kinases (TKs) in the antitumor action of alpha-difluoromethylornithine (DFMO) in breast cancer cells. Breast Cancer Res. Treat. 2008, 112, 255–261. [Google Scholar] [CrossRef]
- Shim, H.J.; Kim, H.I.; Lee, S.T. The associated pyrazolopyrimidines PP1 and PP2 inhibit protein tyrosine kinase 6 activity and suppress breast cancer cell proliferation. Oncol. Lett. 2017, 13, 1463–1469. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Antczak, C.; Mahida, J.P.; Bhinder, B.; Calder, P.A.; Djaballah, H. A high-content biosensor-based screen identifies cell-permeable activators and inhibitors of EGFR function: Implications in drug discovery. J. Biomol. Screen. 2012, 17, 885–899. [Google Scholar] [CrossRef]
- Ye, Q.; Hickey, J.; Summers, K.; Falatovich, B.; Gencheva, M.; Eubank, T.D.; Ivanov, A.V.; Guo, N.L. Multi-Omics Immune Interaction Networks in Lung Cancer Tumorigenesis, Proliferation, and Survival. Int. J. Mol. Sci. 2022, 23, 14978. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef]
- Qian, Y.; Liu, K.J.; Chen, Y.; Flynn, D.C.; Castranova, V.; Shi, X. Cdc42 regulates arsenic-induced NADPH oxidase activation and cell migration through actin filament reorganization. J. Biol. Chem. 2005, 280, 3875–3884. [Google Scholar] [CrossRef]
- Witten, I.H.; Frank, E. Data Mining: Practical Machine Learning Tools and Techniques, 2nd ed.; Morgan Kaufmann: Burlington, MA, USA, 2005. [Google Scholar]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Akritas, M.G. Nearest neighbor estimation of a bivariate distribution under random censoring. Annu. Stat. 1994, 22, 1299–1327. [Google Scholar] [CrossRef]
- Dempster, J.M.R.J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Goncalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e316. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Mohamed, R.; Dukhlallah, D.; Gencheva, M.; Hu, G.; Pearce, M.C.; Kolluri, S.K.; Marsh, C.B.; Eubank, T.D.; Ivanov, A.V.; et al. Molecular Analysis of ZNF71 KRAB in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3752. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Singh, S.; Qian, P.R.; Guo, N.L. Immune-Omics Networks of CD27, PD1, and PDL1 in Non-Small Cell Lung Cancer. Cancers 2021, 13, 4296. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Resistant | Sensitive |
|---|---|---|
| 5-fluorouracil | HSP90AB1, INPPL1, TXNRD1, FGF2 | DDOST |
| alpelisib | FGF2, PSMC3IP, RAD50, S100P | PLSCR1, SEH1L, SMARCD2, PBX2, S100P |
| capecitabine | HSP90AB1, MCM2 | |
| carboplatin | MAP2K2 | |
| cisplatin | PDGFRA, PSMC3IP | DDOST, FGF2, PLSCR1, SLC25A5, SMARCD2, MCM2 |
| cyclophosphamide | FGF2, MCM2 | MCM2 |
| docetaxel | INPPL1, IRF5, RAD52, SMARCD2 | RPUSD2, S100P, SEH1L, SLC25A5 |
| doxorubicin | PDGFRA, PLSCR1, PSMC3IP, RPUSD2, SEH1L, SLC25A5, MCM2 | |
| fulvestrant | TXNRD1, FGF2, S100P | ACOT4, FGF2, INSM1, MCM2, RPUSD2, S100P, SMARCD2 |
| gemcitabine | ACOT4, FGF2, INSM1, SMARCD2 | MAP2K2, PSMC3IP, S100P, SEH1L, SLC25A5, TXNRD1 |
| ixabepilone | DDOST, PBX2, PDGFRA, PLSCR1, RPUSD2 | |
| lapatinib | INPPL1, INSM1, PSMC3IP, S100P, SSBP1 | SMARCD2, PBX2, S100P |
| methotrexate | HSP90AB1, MAP2K2, PBX2, RAD52 | |
| neratinib | PSMC3IP, RAD50, S100P, SLC25A5, TXNRD1 | PBX2 |
| olaparib | FGF2 | FGF2, MAP2K2, PDGFRA, RAD52,S100P, SEH1L, SLC25A5, SSBP1, TXNRD1, |
| paclitaxel | SMARCD2, SSBP1, FGF2, MCM2 | SEH1L, SLC25A5 |
| tucatinib | DDOST, PSMC3IP | FGF2, PBX2, RAD52 |
| vinorelbine | INPPL1 | DDOST, PBX2, PLSCR1, PSMC3IP, RPUSD2, S100P, SEH1L, SLC25A5, SSBP1, TXNRD1 |
| Drug | Resistant Genes | Sensitive Genes |
|---|---|---|
| dasatinib | ACHE, AP3B2, CLRN3, GDAP1, LOXL1, MTMR6, PLCB1, RAB6B, TIGAR | CNTN1, DHRS3, ERN2, GATA6, LRP1B, TDRP |
| PP-2 | PALM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Wang, J.; Ducatman, B.; Raese, R.A.; Rogers, J.L.; Wan, Y.-W.; Dong, C.; Padden, L.; Pugacheva, E.N.; Qian, Y.; et al. Expression-Based Diagnosis, Treatment Selection, and Drug Development for Breast Cancer. Int. J. Mol. Sci. 2023, 24, 10561. https://doi.org/10.3390/ijms241310561
Ye Q, Wang J, Ducatman B, Raese RA, Rogers JL, Wan Y-W, Dong C, Padden L, Pugacheva EN, Qian Y, et al. Expression-Based Diagnosis, Treatment Selection, and Drug Development for Breast Cancer. International Journal of Molecular Sciences. 2023; 24(13):10561. https://doi.org/10.3390/ijms241310561
Chicago/Turabian StyleYe, Qing, Jiajia Wang, Barbara Ducatman, Rebecca A. Raese, Jillian L. Rogers, Ying-Wooi Wan, Chunlin Dong, Lindsay Padden, Elena N. Pugacheva, Yong Qian, and et al. 2023. "Expression-Based Diagnosis, Treatment Selection, and Drug Development for Breast Cancer" International Journal of Molecular Sciences 24, no. 13: 10561. https://doi.org/10.3390/ijms241310561
APA StyleYe, Q., Wang, J., Ducatman, B., Raese, R. A., Rogers, J. L., Wan, Y.-W., Dong, C., Padden, L., Pugacheva, E. N., Qian, Y., & Guo, N. L. (2023). Expression-Based Diagnosis, Treatment Selection, and Drug Development for Breast Cancer. International Journal of Molecular Sciences, 24(13), 10561. https://doi.org/10.3390/ijms241310561