

LncRNAs and CircRNAs in Endoplasmic Reticulum Stress: A Promising Target for Cardiovascular Disease?

 ,
,  and
and
Abstract
1. Introduction
2. ERS and UPR Signalling
3. Role of ERS and UPR in Cardiovascular Diseases
4. Impact of LncRNAs and CircRNAs in ERS and UPR Response on Cardiovascular Diseases
4.1. Atherosclerosis
4.2. Myocardial Infarction
4.3. Cardiac Hypertrophy and Heart Failure
4.4. Dilated Cardiomyopathy
4.5. Diabetic Cardiomyopathy
4.6. Limitations and Unresolved Issues
5. Future Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- English, A.R.; Zurek, N.; Voeltz, G.K. Peripheral ER Structure and Function. Curr. Opin. Cell Biol. 2009, 21, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell. Mol. Life Sci. 2015, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Namba, T. Regulation of Endoplasmic Reticulum Functions. Aging 2015, 7, 901–902. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J.; Bulleid, N.J. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 2020, 9, 1994. [Google Scholar] [CrossRef] [PubMed]
- Bulleid, N.J. Disulfide Bond Formation in the Mammalian Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2012, 4, a013219. [Google Scholar] [CrossRef]
- Braakman, I.; Bulleid, N.J. Protein Folding and Modification in the Mammalian Endoplasmic Reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid Degradation of a Large Fraction of Newly Synthesized Proteins by Proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Rao, R.V.; Bredesen, D.E. Misfolded Proteins, Endoplasmic Reticulum Stress and Neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef]
- Gariballa, N.; Ali, B.R. Endoplasmic Reticulum Associated Protein Degradation (ERAD) in the Pathology of Diseases Related to TGFβ Signaling Pathway: Future Therapeutic Perspectives. Front. Mol. Biosci. 2020, 7, 575608. [Google Scholar] [CrossRef]
- Reggiori, F.; Molinari, M. ER-Phagy: Mechanisms, Regulation, and Diseases Connected to the Lysosomal Clearance of the Endoplasmic Reticulum. Physiol. Rev. 2022, 102, 1393–1448. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of Autophagy by Canonical and Non-Canonical ER Stress Responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Lopata, A.; Kniss, A.; Löhr, F.; Rogov, V.V.; Dötsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369. [Google Scholar] [CrossRef] [PubMed]
- Ferro-Novick, S.; Reggiori, F.; Brodsky, J.L. ER-Phagy, ER Homeostasis, and ER Quality Control: Implications for Disease. Trends Biochem. Sci. 2021, 46, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, A.; Zito, E. ER Stress as a Trigger of UPR and ER-Phagy in Cancer Growth and Spread. Front. Oncol. 2022, 12, 997235. [Google Scholar] [CrossRef] [PubMed]
- Littlejohns, B.; Heesom, K.; Angelini, G.D.; Suleiman, M.S. The Effect of Disease on Human Cardiac Protein Expression Profiles in Paired Samples from Right and Left Ventricles. Clin. Proteom. 2014, 11, 34. [Google Scholar] [CrossRef]
- Ridker, P.M. C-Reactive Protein, Inflammation, and Cardiovascular Disease: Clinical Update. Texas Heart Inst. J. 2005, 32, 384–386. [Google Scholar]
- Wu, L.; Li, H.; Li, X.; Chen, Y.; Zhang, Q.; Cheng, Z.; Fan, Y.; Song, G.; Qian, L. Peptidomic Analysis of Cultured Cardiomyocytes Exposed to Acute Ischemic-Hypoxia. Cell. Physiol. Biochem. 2017, 41, 358–368. [Google Scholar] [CrossRef]
- Vileigas, D.F.; Harman, V.M.; Freire, P.P.; Marciano, C.L.C.; Sant’Ana, P.G.; de Souza, S.L.B.; Mota, G.A.F.; da Silva, V.L.; Campos, D.H.S.; Padovani, C.R.; et al. Landscape of Heart Proteome Changes in a Diet-Induced Obesity Model. Sci. Rep. 2019, 9, 18050. [Google Scholar] [CrossRef]
- Mi, S.; Jiang, H.; Zhang, L.; Xie, Z.; Zhou, J.; Sun, A.; Jin, H.; Ge, J. Regulation of Cardiac-Specific Proteins Expression by Moderate-Intensity Aerobic Exercise Training in Mice With Myocardial Infarction Induced Heart Failure Using MS-Based Proteomics. Front. Cardiovasc. Med. 2021, 8, 1100. [Google Scholar] [CrossRef]
- de Carvalho, A.E.T.S.; Cordeiro, M.A.; Rodrigues, L.S.; Ortolani, D.; Spadari, R.C. Stress-Induced Differential Gene Expression in Cardiac Tissue. Sci. Rep. 2021, 11, 9129. [Google Scholar] [CrossRef]
- Cauwenberghs, N.; Sabovčik, F.; Magnus, A.; Haddad, F.; Kuznetsova, T. Proteomic Profiling for Detection of Early-Stage Heart Failure in the Community. ESC Heart Fail. 2021, 8, 2928–2939. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 Facilitates Heart Development by Antagonizing Oxidative and ER Stress Responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef] [PubMed]
- Bozi, L.H.M.; Takano, A.P.C.; Campos, J.C.; Rolim, N.; Dourado, P.M.M.; Voltarelli, V.A.; Wisløff, U.; Ferreira, J.C.B.; Barreto-Chaves, M.L.M.; Brum, P.C. Endoplasmic Reticulum Stress Impairs Cardiomyocyte Contractility through JNK-Dependent Upregulation of BNIP3. Int. J. Cardiol. 2018, 272, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Burgeiro, A.; Fonseca, A.C.; Espinoza, D.; Carvalho, L.; Lourenço, N.; Antunes, M.; Carvalho, E. Proteostasis in Epicardial versus Subcutaneous Adipose Tissue in Heart Failure Subjects with and without Diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt A, 2183–2198. [Google Scholar] [CrossRef]
- Shimizu, T.; Maruyama, K.; Kawamura, T.; Urade, Y.; Wada, Y. PERK Participates in Cardiac Valve Development via Fatty Acid Oxidation and Endocardial-Mesenchymal Transformation. Sci. Rep. 2020, 10, 20094. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 Activation Confers Global Protection in Widespread Disease Models by Reprograming Cellular Proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef]
- Steiger, D.; Yokota, T.; Li, J.; Ren, S.; Minamisawa, S.; Wang, Y. The Serine/Threonine-Protein Kinase/Endoribonuclease IRE1α Protects the Heart against Pressure Overload-Induced Heart Failure. J. Biol. Chem. 2018, 293, 9652–9661. [Google Scholar] [CrossRef]
- Shimizu, T.; Taguchi, A.; Higashijima, Y.; Takubo, N.; Kanki, Y.; Urade, Y.; Wada, Y. PERK-Mediated Suppression of MicroRNAs by Sildenafil Improves Mitochondrial Dysfunction in Heart Failure. iScience 2020, 23, 101410. [Google Scholar] [CrossRef]
- Glembotski, C.C.; Arrieta, A.; Blackwood, E.A.; Stauffer, W.T. ATF6 as a Nodal Regulator of Proteostasis in the Heart. Front. Physiol. 2020, 11, 267. [Google Scholar] [CrossRef]
- Al-Yacoub, N.; Colak, D.; Mahmoud, S.A.; Hammonds, M.; Muhammed, K.; Al-Harazi, O.; Assiri, A.M.; Al-Buraiki, J.; Al-Habeeb, W.; Poizat, C. Mutation in FBXO32 Causes Dilated Cardiomyopathy through Up-Regulation of ER-Stress Mediated Apoptosis. Commun. Biol. 2021, 4, 884. [Google Scholar] [CrossRef]
- Yao, Y.; Lu, Q.; Hu, Z.; Yu, Y.; Chen, Q.; Wang, Q.K. A Non-Canonical Pathway Regulates ER Stress Signaling and Blocks ER Stress-Induced Apoptosis and Heart Failure. Nat. Commun. 2017, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.I.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged Endoplasmic Reticulum Stress in Hypertrophic and Failing Heart after Aortic Constriction: Possible Contribution of Endoplasmic Reticulum Stress to Cardiac Myocyte Apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, O.; Higuchi, Y.; Hirotani, S.; Kashiwase, K.; Nakayama, H.; Hikoso, S.; Takeda, T.; Watanabe, T.; Asahi, M.; Taniike, M.; et al. Targeted Deletion of Apoptosis Signal-Regulating Kinase 1 Attenuates Left Ventricular Remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 15883–15888. [Google Scholar] [CrossRef]
- Zhou, Y.; Murugan, D.D.; Khan, H.; Huang, Y.; Cheang, W.S. Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases. Antioxidants 2021, 10, 1167. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic Reticulum Stress in the Heart: Insights into Mechanisms and Drug Targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER Stress in Cardiovascular Disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cardiovascular Diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 8, 758311. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A Guide to Understanding Endoplasmic Reticulum Stress in Metabolic Disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 Is Essential for Endoplasmic Reticulum Stress-Induced Neuronal Cell Death Triggered by Expanded Polyglutamine Repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Hillary, R.F.; FitzGerald, U. A Lifetime of Stress: ATF6 in Development and Homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/P62 Protein Is Associated with Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.T.; Darko, C.; Sharma, R.B.; Alonso, L.C. Endoplasmic Reticulum Stress Induced Proliferation Remains Intact in Aging Mouse β-Cells. Front. Endocrinol. 2021, 12, 734079. [Google Scholar] [CrossRef] [PubMed]
- Legg, K. Defective UPR Linked to β-Cell Dedifferentiation. Nat. Rev. Endocrinol. 2022, 18, 716. [Google Scholar] [CrossRef] [PubMed]
- Turishcheva, E.; Vildanova, M.; Onishchenko, G.; Smirnova, E. The Role of Endoplasmic Reticulum Stress in Differentiation of Cells of Mesenchymal Origin. Biochemistry 2022, 87, 916–931. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sudo, T.; Morishima, N. Endoplasmic Reticulum Stress Signaling Transmitted by ATF6 Mediates Apoptosis during Muscle Development. J. Cell Biol. 2005, 169, 555–560. [Google Scholar] [CrossRef]
- Gallot, Y.S.; Bohnert, K.R.; Straughn, A.R.; Xiong, G.; Hindi, S.M.; Kumar, A. PERK Regulates Skeletal Muscle Mass and Contractile Function in Adult Mice. FASEB J. 2019, 33, 1946–1962. [Google Scholar] [CrossRef]
- Tokutake, Y.; Yamada, K.; Hayashi, S.; Arai, W.; Watanabe, T.; Yonekura, S. IRE1-XBP1 Pathway of the Unfolded Protein Response Is Required during Early Differentiation of C2C12 Myoblasts. Int. J. Mol. Sci. 2019, 21, 182. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Tomaz da Silva, M.; Bhat, R.; Bohnert, K.R.; Iwawaki, T.; Kumar, A. The IRE1/XBP1 Signaling Axis Promotes Skeletal Muscle Regeneration through a Cell Non-Autonomous Mechanism. Elife 2021, 10, e73215. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Hindi, S.M.; Mann, A.K.; Gallot, Y.S.; Bohnert, K.R.; Cavener, D.R.; Whittemore, S.R.; Kumar, A. The PERK Arm of the Unfolded Protein Response Regulates Satellite Cell-Mediated Skeletal Muscle Regeneration. Elife 2017, 6, e22871. [Google Scholar] [CrossRef] [PubMed]
- Mesbah Moosavi, Z.S.; Hood, D.A. The Unfolded Protein Response in Relation to Mitochondrial Biogenesis in Skeletal Muscle Cells. Am. J. Physiol. Cell Physiol. 2017, 312, C583–C594. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The Aftermath of the Interplay between the Endoplasmic Reticulum Stress Response and Redox Signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of Calcium Homeostasis and Flux between the Endoplasmic Reticulum and the Cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The Role of the Unfolded Protein Response in Cancer Progression: From Oncogenesis to Chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Aghaei, M.; Dastghaib, S.; Aftabi, S.; Aghanoori, M.-R.; Alizadeh, J.; Mokarram, P.; Mehrbod, P.; Ashrafizadeh, M.; Zarrabi, A.; McAlinden, K.D.; et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life 2020, 11, 1. [Google Scholar] [CrossRef]
- Mokarram, P.; Albokashy, M.; Zarghooni, M.; Moosavi, M.A.; Sepehri, Z.; Chen, Q.M.; Hudecki, A.; Sargazi, A.; Alizadeh, J.; Moghadam, A.R.; et al. New Frontiers in the Treatment of Colorectal Cancer: Autophagy and the Unfolded Protein Response as Promising Targets. Autophagy 2017, 13, 781–819. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER Stress and UPR in Alzheimer’s Disease: Mechanisms, Pathogenesis, Treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef]
- Steinberger, A.E.; Tecos, M.E.; Phelps, H.M.; Rubin, D.C.; Davidson, N.O.; Guo, J.; Warner, B.W. A Novel Maladaptive Unfolded Protein Response as a Mechanism for Small Bowel Resection-Induced Liver Injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G165–G176. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-Mediated Precision Autophagy Targets Cytoplasmic Regulators of Innate Immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xie, J.; Wang, Y.-Z.; Gan, Y.-R.; Wei, L.; Ding, G.-W.; Ding, Y.-H.; Xie, D.-X. Overexpression of LncRNA Dancr Inhibits Apoptosis and Enhances Autophagy to Protect Cardiomyocytes from Endoplasmic Reticulum Stress Injury via Sponging MicroRNA-6324. Mol. Med. Rep. 2021, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Nakatogawa, H. ER-Phagy: Selective Autophagy of the Endoplasmic Reticulum. EMBO Rep. 2022, 23, e55192. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-Mediated Removal of IRE1α Drives Crohn’s Disease-like Ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Ji, C.H.; Kim, H.Y.; Heo, A.J.; Lee, S.H.; Lee, M.J.; Kim, S.B.; Srinivasrao, G.; Mun, S.R.; Cha-Molstad, H.; Ciechanover, A.; et al. The N-Degron Pathway Mediates ER-Phagy. Mol. Cell 2019, 75, 1058–1072.e9. [Google Scholar] [CrossRef]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature Ribosomes Are Selectively Degraded upon Starvation by an Autophagy Pathway Requiring the Ubp3p/Bre5p Ubiquitin Protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Ossareh-Nazari, B.; Bonizec, M.; Cohen, M.; Dokudovskaya, S.; Delalande, F.; Schaeffer, C.; Van Dorsselaer, A.; Dargemont, C. Cdc48 and Ufd3, New Partners of the Ubiquitin Protease Ubp3, Are Required for Ribophagy. EMBO Rep. 2010, 11, 548–554. [Google Scholar] [CrossRef]
- Zheng, W.; Xie, W.; Yin, D.; Luo, R.; Liu, M.; Guo, F. ATG5 and ATG7 Induced Autophagy Interplays with UPR via PERK Signaling. Cell Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef]
- Liao, Y.; Duan, B.; Zhang, Y.; Zhang, X.; Xia, B. Excessive ER-Phagy Mediated by the Autophagy Receptor FAM134B Results in ER Stress, the Unfolded Protein Response, and Cell Death in HeLa Cells. J. Biol. Chem. 2019, 294, 20009–20023. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Yang, D.; Yu, X.; Irwin, D.M.; Niu, G.; Tan, H. Excessive Autophagy Activation and Increased Apoptosis Are Associated with Palmitic Acid-Induced Cardiomyocyte Insulin Resistance. J. Diabetes Res. 2017, 2017, 2376893. [Google Scholar] [CrossRef] [PubMed]
- Prola, A.; Nichtova, Z.; Pires Da Silva, J.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic Reticulum Stress Induces Cardiac Dysfunction through Architectural Modifications and Alteration of Mitochondrial Function in Cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Duan, Q.; He, M.; Li, X.; Wang, B.; Zhou, C.; Wu, L.; Wen, Z.; Chen, C.; Wang, D.W.; et al. Ranolazine Prevents Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure by Restoring Aberrant Na(+) and Ca(2+) Handling. J. Cell. Physiol. 2019, 234, 11587–11601. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kwak, D.; Lu, Z.; Xu, X.; Fassett, J.; Wang, H.; Wei, Y.; Cavener, D.R.; Hu, X.; Hall, J.; et al. Endoplasmic Reticulum Stress Sensor Protein Kinase R-like Endoplasmic Reticulum Kinase (PERK) Protects against Pressure Overload-Induced Heart Failure and Lung Remodeling. Hypertension 2014, 64, 738–744. [Google Scholar] [CrossRef]
- Jin, J.-K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-Box Binding Protein 1 Couples the Unfolded Protein Response to Hexosamine Biosynthetic Pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP Homologous Protein Attenuates Endoplasmic Reticulum-Mediated Apoptosis and Cardiac Dysfunction Induced by Pressure Overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Völkers, M.; Thuerauf, D.J.; Khan, M.; Mohsin, S.; Respress, J.L.; Wang, W.; Gude, N.; Müller, O.J.; Wehrens, X.H.T.; et al. Hrd1 and ER-Associated Protein Degradation, ERAD, Are Critical Elements of the Adaptive ER Stress Response in Cardiac Myocytes. Circ. Res. 2015, 117, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wu, M.; Li, X.; Zhao, R.; Zhao, Y.; Liu, L.; Wang, S. Role of Endoplasmic Reticulum Stress in Atherosclerosis and Its Potential as a Therapeutic Target. Oxid. Med. Cell. Longev. 2020, 2020, 9270107. [Google Scholar] [CrossRef]
- Hu, J.; Huang, C.-X.; Rao, P.-P.; Cao, G.-Q.; Zhang, Y.; Zhou, J.-P.; Zhu, L.-Y.; Liu, M.-X.; Zhang, G.-G. MicroRNA-155 Inhibition Attenuates Endoplasmic Reticulum Stress-Induced Cardiomyocyte Apoptosis Following Myocardial Infarction via Reducing Macrophage Inflammation. Eur. J. Pharmacol. 2019, 857, 172449. [Google Scholar] [CrossRef]
- Toro, R.; Pérez-Serra, A.; Mangas, A.; Campuzano, O.; Sarquella-Brugada, G.; Quezada-Feijoo, M.; Ramos, M.; Alcalá, M.; Carrera, E.; García-Padilla, C.; et al. MiR-16-5p Suppression Protects Human Cardiomyocytes against Endoplasmic Reticulum and Oxidative Stress-Induced Injury. Int. J. Mol. Sci. 2022, 23, 1036. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Villa, E.; Bonet, F.; Hernandez-Torres, F.; Campuzano, Ó.; Sarquella-Brugada, G.; Quezada-Feijoo, M.; Ramos, M.; Mangas, A.; Toro, R. The Role of MicroRNAs in Dilated Cardiomyopathy: New Insights for an Old Entity. Int. J. Mol. Sci. 2022, 23, 3573. [Google Scholar] [CrossRef] [PubMed]
- Demirel-Yalciner, T.; Sozen, E.; Ozer, N.K. Endoplasmic Reticulum Stress and MiRNA Impairment in Aging and Age-Related Diseases. Front. Aging 2021, 2, 790702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Jiang, S.; Wu, N.; Shi, E.; Yang, L.; Li, Q. MiR-17-5p-Mediated Endoplasmic Reticulum Stress Promotes Acute Myocardial Ischemia Injury through Targeting Tsg101. Cell Stress Chaperones 2021, 26, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, F.C.; Werner, A.; John, D.; Boeckel, J.-N.; Melissari, M.-T.; Grote, P.; Glaser, S.F.; Demolli, S.; Uchida, S.; Michalik, K.M.; et al. Identification and Functional Characterization of Hypoxia-Induced Endoplasmic Reticulum Stress Regulating LncRNA (HypERlnc) in Pericytes. Circ. Res. 2017, 121, 368–375. [Google Scholar] [CrossRef]
- Li, X.; Zhao, J.; Geng, J.; Chen, F.; Wei, Z.; Liu, C.; Zhang, X.; Li, Q.; Zhang, J.; Gao, L.; et al. Long Non-Coding RNA MEG3 Knockdown Attenuates Endoplasmic Reticulum Stress-Mediated Apoptosis by Targeting P53 Following Myocardial Infarction. J. Cell. Mol. Med. 2019, 23, 8369–8380. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective Autophagy of Intracellular Organelles: Recent Research Advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Chen, J.; Hu, Q.; Zhang, B.-F.; Liu, X.-P.; Yang, S.; Jiang, H. Long Noncoding RNA UCA1 Inhibits Ischaemia/Reperfusion Injury Induced Cardiomyocytes Apoptosis via Suppression of Endoplasmic Reticulum Stress. Genes Genom. 2019, 41, 803–810. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, Y.; Han, B.; Yang, L.; Chen, X.; Huang, R.; Wu, F.; Chao, J.; Liu, P.; Hu, G.; et al. Circular RNA DLGAP4 Ameliorates Ischemic Stroke Outcomes by Targeting MiR-143 to Regulate Endothelial-Mesenchymal Transition Associated with Blood-Brain Barrier Integrity. J. Neurosci. 2018, 38, 32–50. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, M.; Yu, X.; Shui, X.; Tang, L.; Chen, Z.; Xiong, Z. LncRNA NBR2 Attenuates Angiotensin II-Induced Myocardial Hypertrophy through Repressing ER Stress via Activating LKB1/AMPK/Sirt1 Pathway. Bioengineered 2022, 13, 13667–13679. [Google Scholar] [CrossRef]
- Qiu, Z.; Chen, W.; Liu, Y.; Jiang, B.; Yin, L.; Chen, X. LncRNA AC061961.2 Overexpression Inhibited Endoplasmic Reticulum Stress Induced Apoptosis in Dilated Cardiomyopathy Rats and Cardiomyocytes via Activating Wnt/β-Catenin Pathway. J. Recept. Signal Transduct. Res. 2021, 41, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Duan, J.; Liao, J.; Wang, Y.; Xiao, X.; Li, L.; Liu, Y.; Gu, H.; Yang, P.; Fu, D.; et al. LncRNA H19 Inhibits ER Stress Induced Apoptosis and Improves Diabetic Cardiomyopathy by Regulating PI3K/AKT/MTOR Axis. Aging 2022, 14, 6809–6828. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-H.; Xia, N.; Zhou, S.-F.; Tang, T.-T.; Yan, X.-X.; Lv, B.-J.; Nie, S.-F.; Wang, J.; Iwakura, Y.; Xiao, H.; et al. Interleukin-17A Contributes to Myocardial Ischemia/Reperfusion Injury by Regulating Cardiomyocyte Apoptosis and Neutrophil Infiltration. J. Am. Coll. Cardiol. 2012, 59, 420–429. [Google Scholar] [CrossRef]
- Bo, Z.; Huang, S.; Li, L.; Chen, L.; Chen, P.; Luo, X.; Shi, F.; Zhu, B.; Shen, L. EGR2 Is a Hub-Gene in Myocardial Infarction and Aggravates Inflammation and Apoptosis in Hypoxia-Induced Cardiomyocytes. BMC Cardiovasc. Disord. 2022, 22, 373. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; White, H.D.; Jaffe, A.S.; Apple, F.S.; Galvani, M.; Katus, H.A.; Newby, L.K.; Ravkilde, J.; Chaitman, B.; et al. Universal Definition of Myocardial Infarction. Circulation 2007, 116, 2634–2653. [Google Scholar] [CrossRef]
- Zhu, H.; Zhou, H. Novel Insight into the Role of Endoplasmic Reticulum Stress in the Pathogenesis of Myocardial Ischemia-Reperfusion Injury. Oxid. Med. Cell. Longev. 2021, 2021, 5529810. [Google Scholar] [CrossRef]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.-F.; Dohmen, P.M.; Choi, Y.-H.; Wahlers, T.; et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef]
- Qiu, G.; Ren, L.; Jiang, H.; Shi, X.; Cao, L. Dilated Cardiomyopathy-Related Stroke Mimicking Large-Artery Atherosclerosis-Related Stroke: Report of Two Cases. Signa Vitae 2021, 17, 150–156. [Google Scholar] [CrossRef]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving Concepts in Dilated Cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Suzuki, M.; Yuasa, S.; Mimura, N.; Shinozuka, N.; Takada, Y.; Suzuki, M.; Nishino, T.; Nakaya, H.; Koseki, H.; et al. Dilated Cardiomyopathy Caused by Aberrant Endoplasmic Reticulum Quality Control in Mutant KDEL Receptor Transgenic Mice. Mol. Cell. Biol. 2004, 24, 8007–8017. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
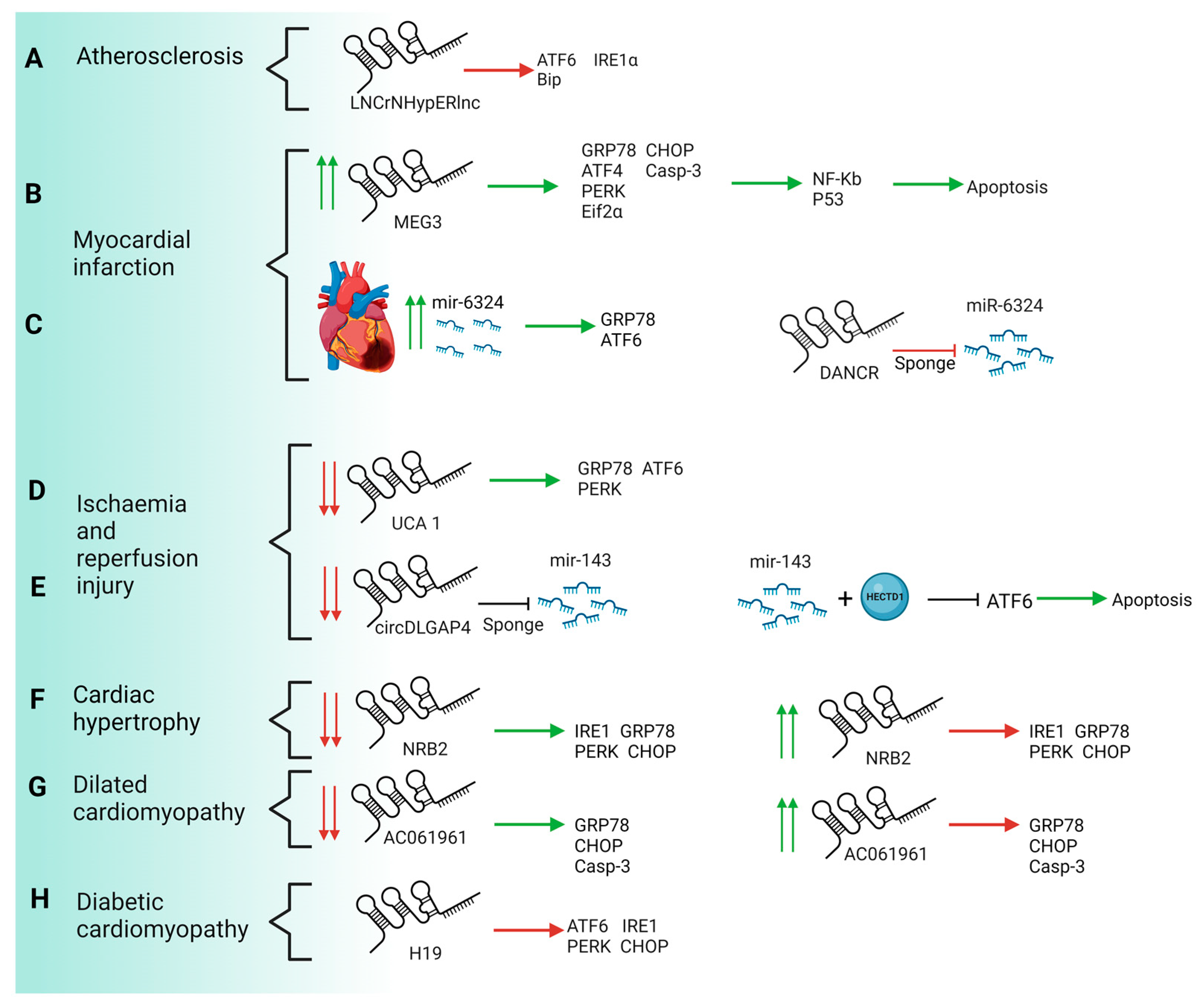
| ncRNA | CVD | Effects | Mechanisms | Subjects and Size | Study Model | Type of the Study | Ref. |
|---|---|---|---|---|---|---|---|
| HypERlnc | Atherosclerosis | Inhibition of maladaptive UPR | Decrease in mRNA/protein levels of ATF6, IRE1α and Bip | Human cardiac tissue from patients with heart failure (HF) and pericytes exposed to hypoxia | Human | Gain and loss function assay ex vivo and in vitro | [85] |
| MEG3 | Myocardial infarction | Activation of ERS-mediated apoptosis | Upregulation of mRNA/protein levels of GRP78, ATF4, PERK, eiF2α, CHOP and caspase 3 | Infarcted hearts and hypoxic neonatal mice ventricular myocytes | Mice | Lost function assay in vitro | [86] |
| DANCR | Myocardial infarction | Inhibition of ERS-mediated apoptosis | Repression of GRP78, Beclin 1, p-IRE1α, p-IRE1α/IRE1α and Xbp1s by sponging miR-6324 | H9C2 cardiomyocytes | Rat | Gain and loss function assay in vitro | [87] |
| UCA1 | Ischaemia and reperfusion injury | Reduction of ROS production and improvement of mitochondrial function | Decrease in GRP78, ATF6 and PERK transcription | H9C2 cardiomyocytes | Rat | Gain and loss function assay in vitro | [88] |
| circDLGAP4 | Ischaemia and reperfusion injury | Repression of ATF6 signalling pathway | Sponge to miR-143 avoiding to repression of HECTD1 | Endothelial cells | Mouse | Gain function assay in vitro | [89] |
| NRB2 | Heart hypertrophy and heart failure | Activation of LKB1/AMPK/Sirt1 pathway. | Decrease in mRNA/protein levels of PERK, IRE1, GRP78 and CHOP | Human cardiomyocytes cell line | Human | Gain-of-function assay in vitro | [90] |
| AC061961.2 | Dilated cardiomyopathy | Reversion of apoptosis by activating Wnt/β-catenin signalling | Decrease in mRNA/protein levels of GRP78, CHOP and caspase 3 | Vitro and in vivo Adriamycin-induced DCM | Rat | Gain-of-function assay in vitro | [91] |
| H19 | Diabetic cardiomyopathy | Repression of cardiomyocyte apoptosis | Decrease in mRNA/protein levels of ATF6, PERK, CHOP and IRE1α | Induced DM mice | Mice | Gain function assay in vitro | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Amaro, F.J.; Garcia-Padilla, C.; Franco, D.; Daimi, H. LncRNAs and CircRNAs in Endoplasmic Reticulum Stress: A Promising Target for Cardiovascular Disease? Int. J. Mol. Sci. 2023, 24, 9888. https://doi.org/10.3390/ijms24129888
Martinez-Amaro FJ, Garcia-Padilla C, Franco D, Daimi H. LncRNAs and CircRNAs in Endoplasmic Reticulum Stress: A Promising Target for Cardiovascular Disease? International Journal of Molecular Sciences. 2023; 24(12):9888. https://doi.org/10.3390/ijms24129888
Chicago/Turabian StyleMartinez-Amaro, Francisco José, Carlos Garcia-Padilla, Diego Franco, and Houria Daimi. 2023. "LncRNAs and CircRNAs in Endoplasmic Reticulum Stress: A Promising Target for Cardiovascular Disease?" International Journal of Molecular Sciences 24, no. 12: 9888. https://doi.org/10.3390/ijms24129888
APA StyleMartinez-Amaro, F. J., Garcia-Padilla, C., Franco, D., & Daimi, H. (2023). LncRNAs and CircRNAs in Endoplasmic Reticulum Stress: A Promising Target for Cardiovascular Disease? International Journal of Molecular Sciences, 24(12), 9888. https://doi.org/10.3390/ijms24129888