
Differential Lipidomics, Metabolomics and Immunological Analysis of Alcoholic and Non-Alcoholic Steatohepatitis in Mice

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
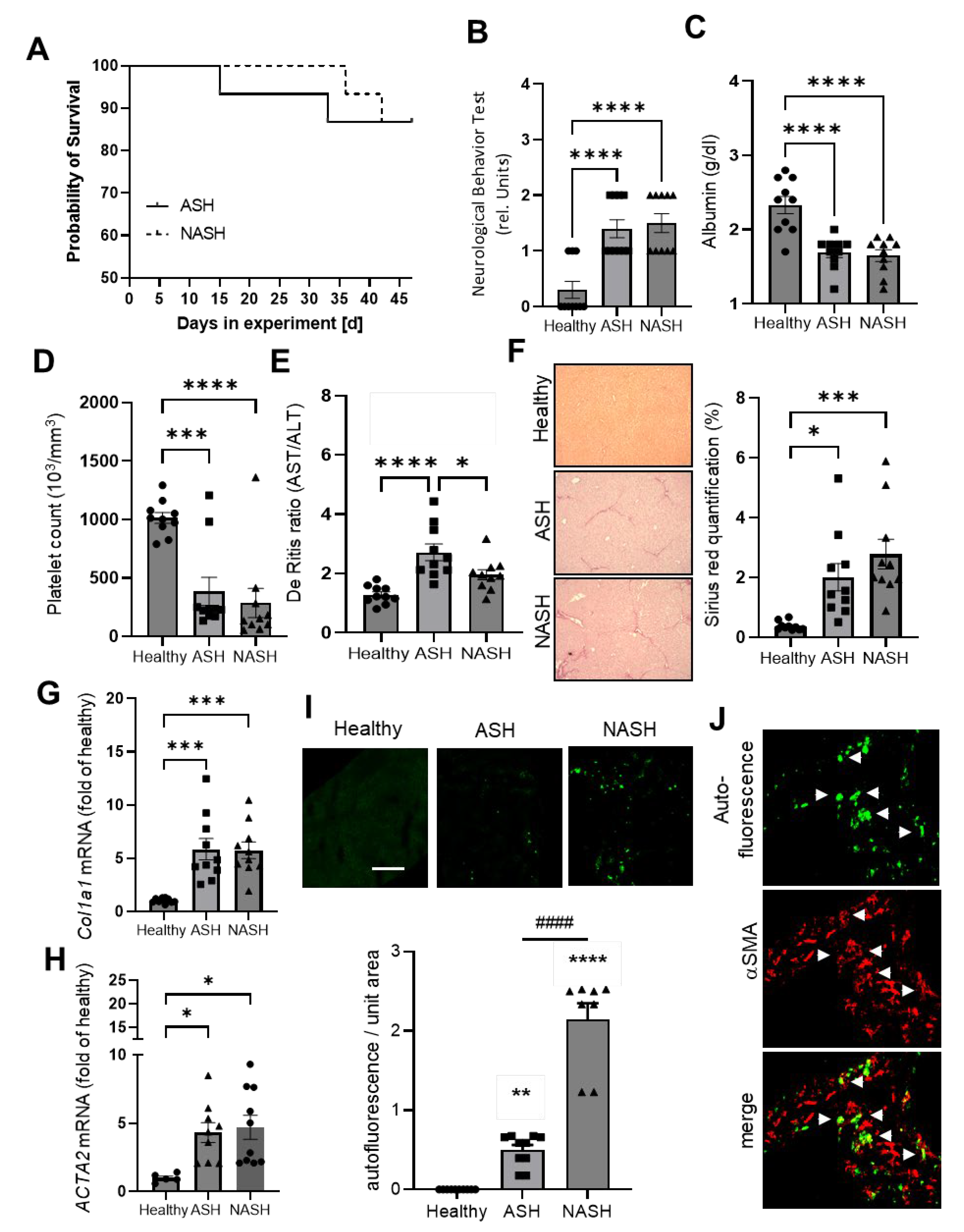
2.1. Disease Severity in Mice Models for ASH and NASH Is Comparable
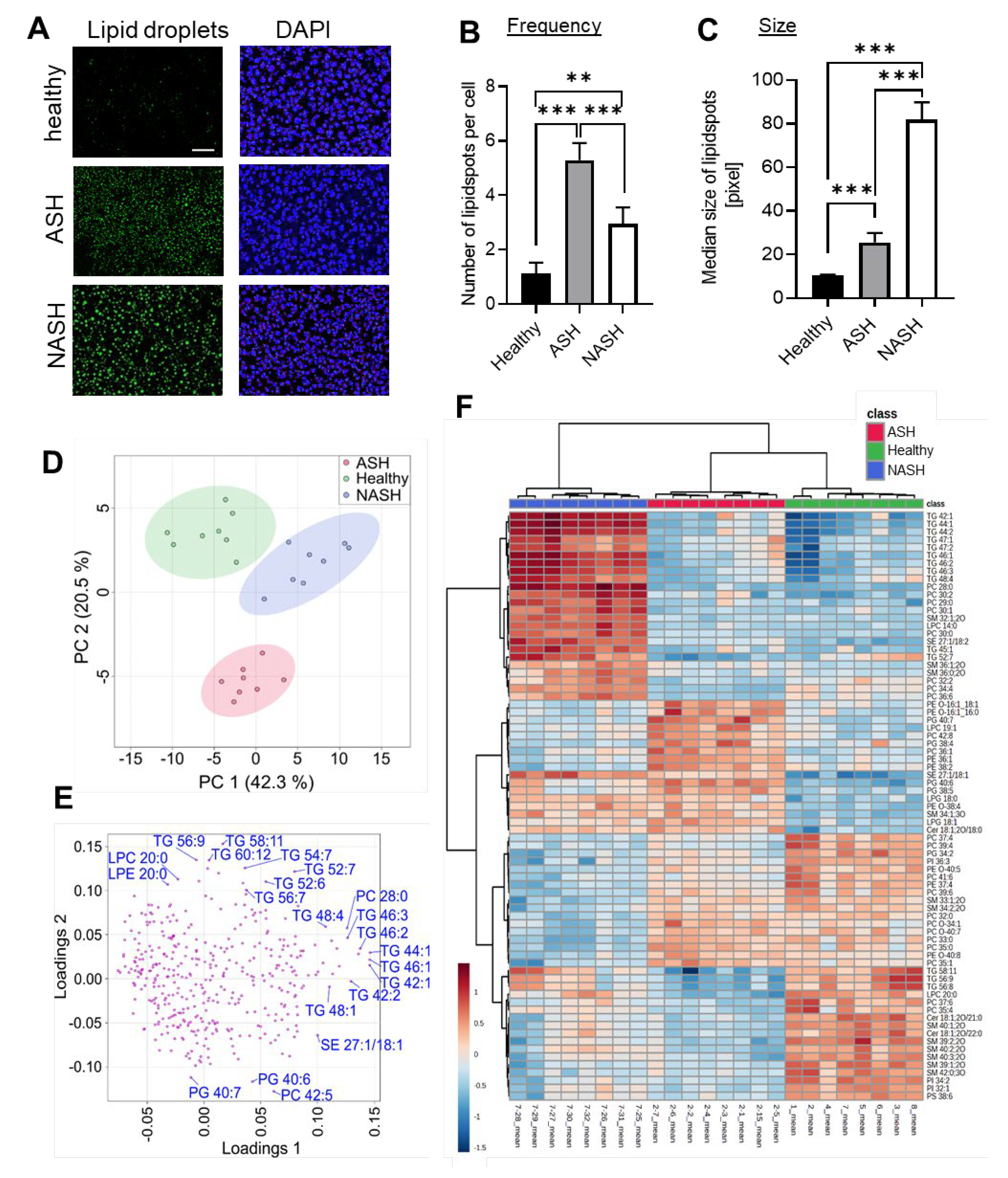
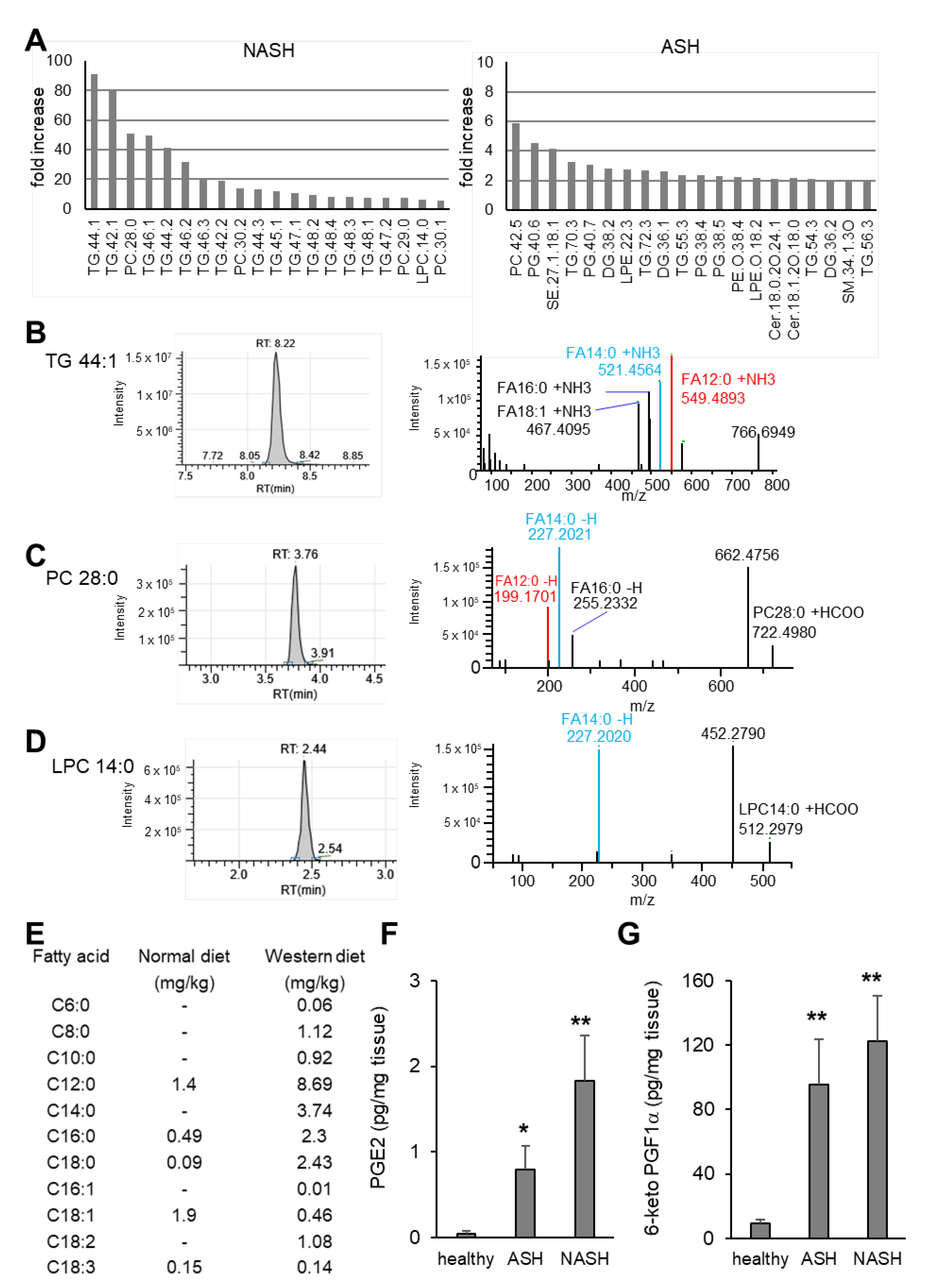
2.2. Major Changes in the Hepatic Lipid Content Are Influenced by the Specialized Diets
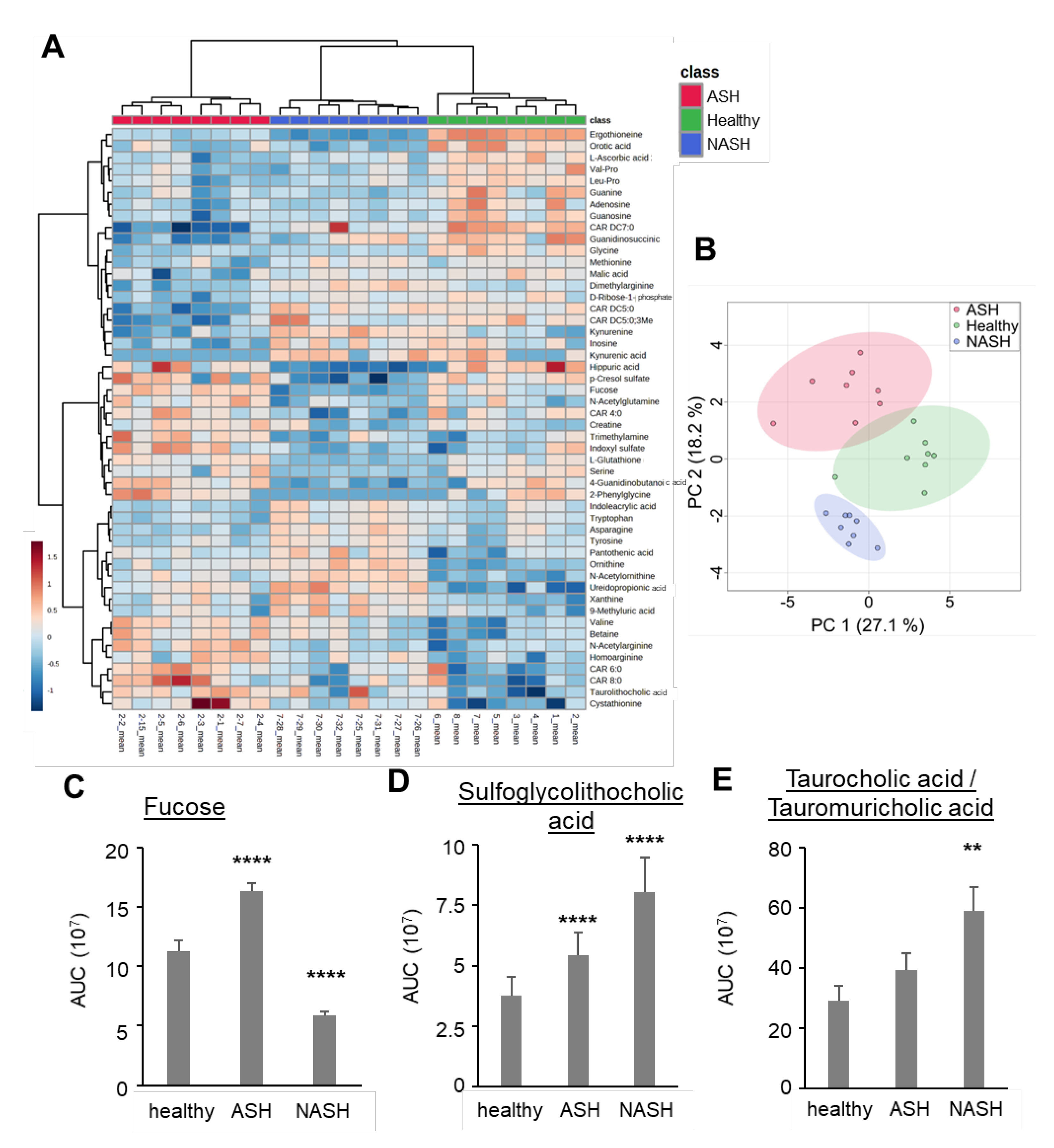
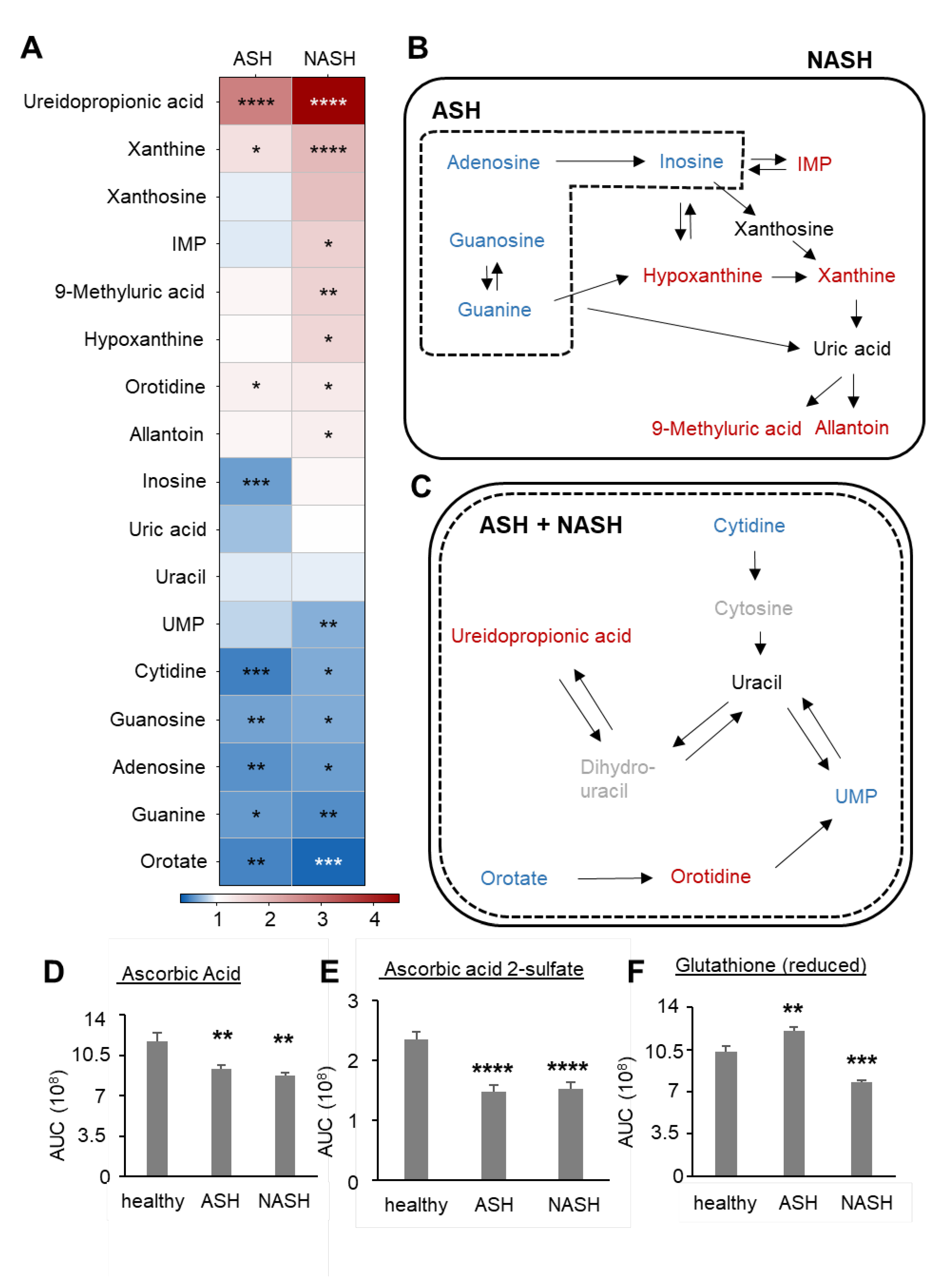
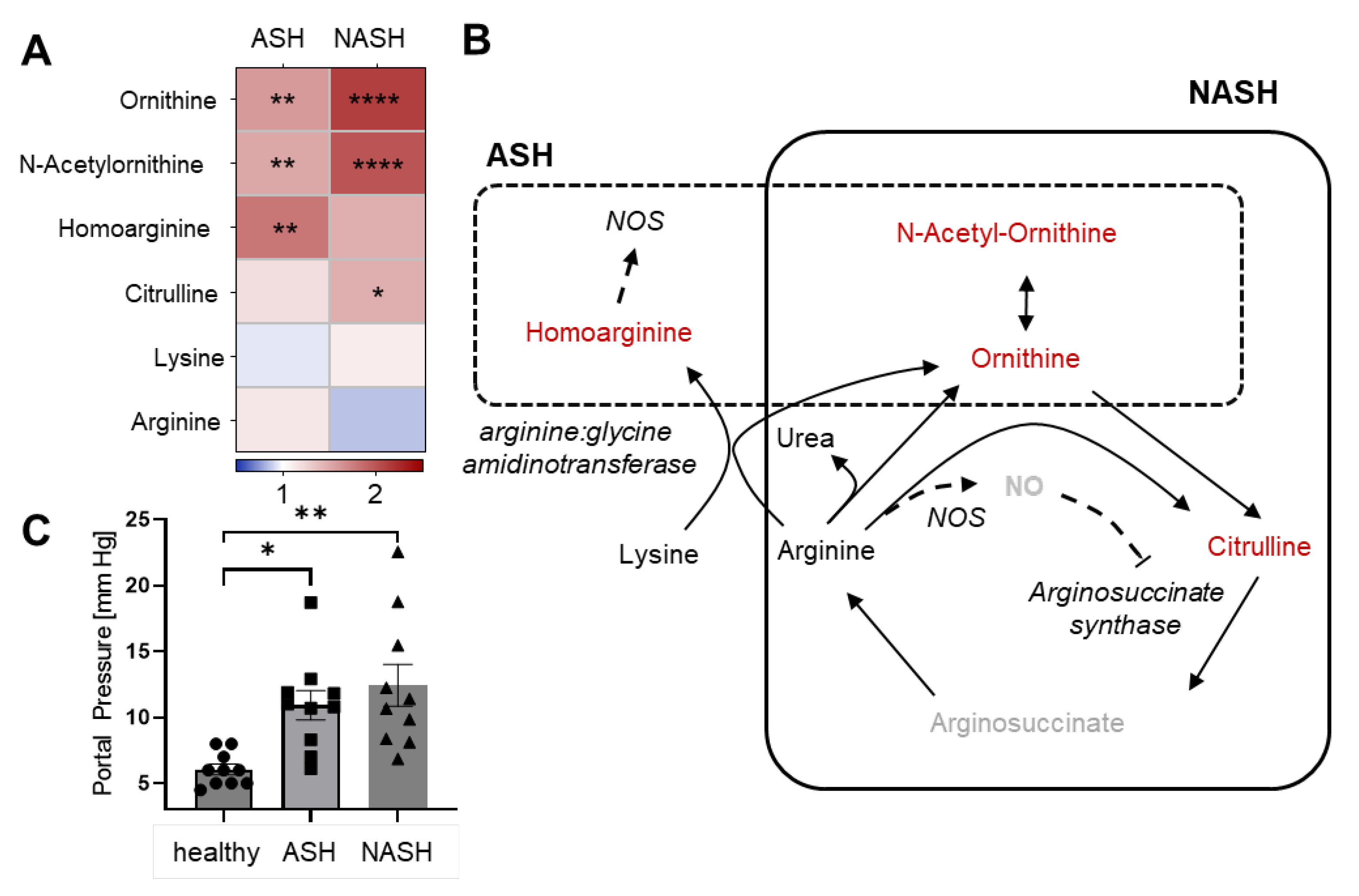
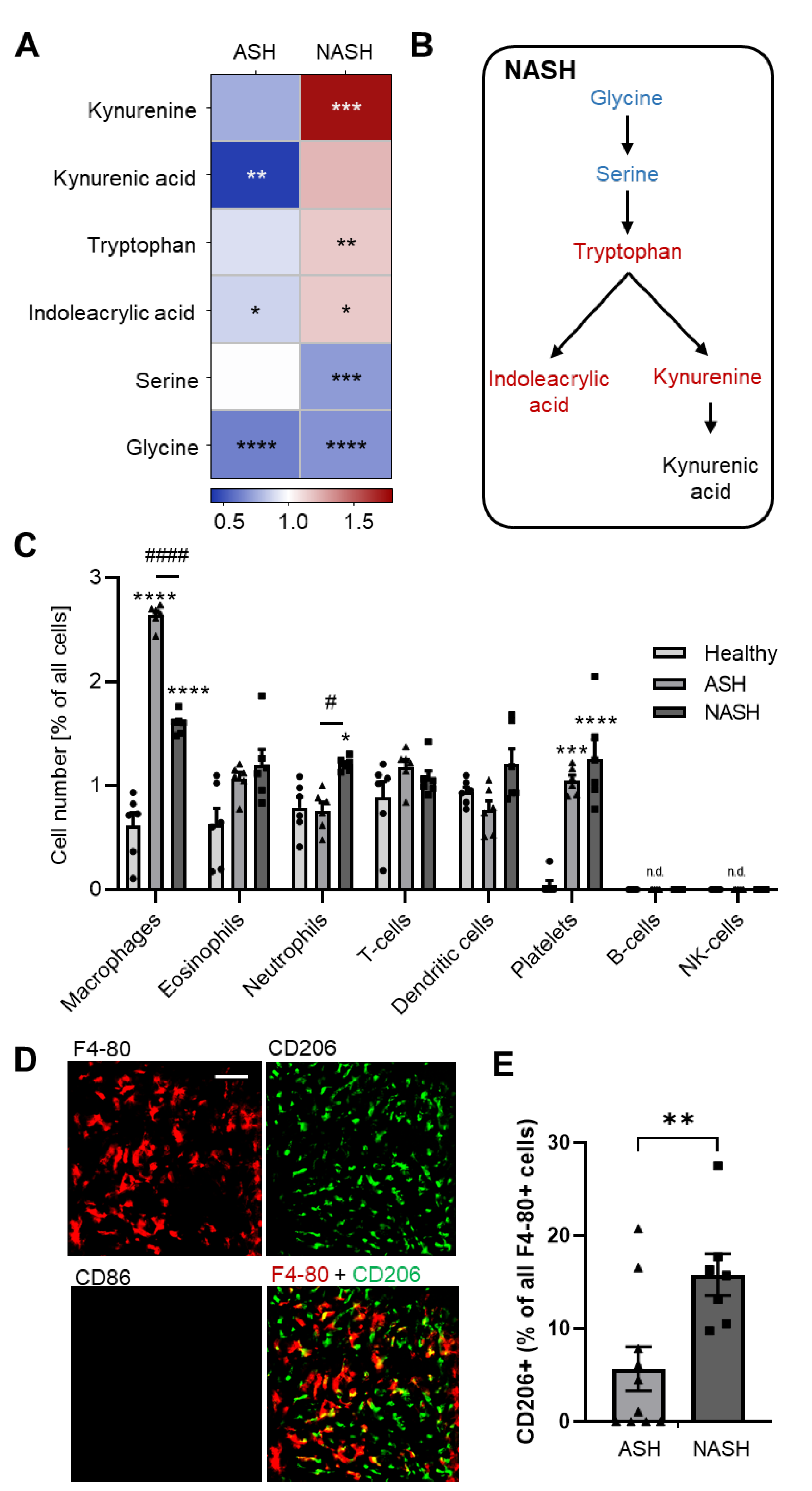
2.3. Metabolomics Reveal Disease-Specific Responses in ASH and NASH
2.4. Hepatic Immune Responses Differ between ASH and NASH
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Induction of Chronic Liver Disease
4.3. Brain Injury Assessment
4.4. Blood Parameters
4.5. Sirius Red Staining
4.6. Quantitative Real-Time PCR
4.7. Portal Pressure Measurement
4.8. High-Content Multiplex Imaging
4.9. Image Analysis
4.10. Lipid Droplet Quantification
4.11. Profiling of Lipids and Polar Metabolites
4.12. Oxylipine Analysis
4.13. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naghavi, M.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Adetokunboh, O.; Afshin, A.; Agrawal, A.; et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Kim, S.-J.; Gao, B. Alcohol, adipose tissue and liver disease: Mechanistic links and clinical considerations. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Molina, P.E. Cellular Bioenergetics: Experimental Evidence for Alcohol-induced Adaptations. Function 2022, 3, zqac039. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef]
- Bailey, S.M.; Cunningham, C.C. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic. Biol. Med. 2002, 32, 11–16. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Brol, M.J.; Rösch, F.; Schierwagen, R.; Magdaleno, F.; Uschner, F.E.; Manekeller, S.; Queck, A.; Schwarzkopf, K.; Odenthal, M.; Drebber, U.; et al. Combination of CCl4 with alcoholic and metabolic injuries mimics human liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G182–G194. [Google Scholar] [CrossRef]
- Kamm, D.R.; McCommis, K.S. Hepatic stellate cells in physiology and pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Kimura, K.; Maezawa, Y.; Ohata, M.; Mizuhara, Y.; Hirakawa, J.; Nakajima, H.; Toda, G. Urinary level of L-fucose as a marker of alcoholic liver disease. Alcohol. Clin. Exp. Res. 1993, 17, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Niu, M.; Tang, W.; Hu, J.; Wei, G.; He, Z.; Chen, Y.; Jiang, Y.; Chen, P. L-Fucose ameliorates high-fat diet-induced obesity and hepatic steatosis in mice. J. Transl. Med. 2018, 16, 344. [Google Scholar] [CrossRef]
- Hao, G.; Xie, L.; Gross, S.S. Argininosuccinate synthetase is reversibly inactivated by S-nitrosylation in vitro and in vivo. J. Biol. Chem. 2004, 279, 36192–36200. [Google Scholar] [CrossRef]
- Tsikas, D.; Wu, G. Homoarginine, arginine, and relatives: Analysis, metabolism, transport, physiology, and pathology. Amino Acids 2015, 47, 1697–1702. [Google Scholar] [CrossRef]
- Tsuji, A.; Ikeda, Y.; Yoshikawa, S.; Taniguchi, K.; Sawamura, H.; Morikawa, S.; Nakashima, M.; Asai, T.; Matsuda, S. The Tryptophan and Kynurenine Pathway Involved in the Development of Immune-Related Diseases. Int. J. Mol. Sci. 2023, 24, 5742. [Google Scholar] [CrossRef]
- Kolbinger, A.; Schäufele, T.J.; Steigerwald, H.; Friedel, J.; Pierre, S.; Geisslinger, G.; Scholich, K. Eosinophil-derived IL-4 is necessary to establish the inflammatory structure in innate inflammation. EMBO Mol. Med. 2023, 15, e16796. [Google Scholar] [CrossRef]
- Kolbinger, A.; Kestner, R.I.; Jencio, L.; Schäufele, T.J.; Vutukuri, R.; Pfeilschifter, W.; Scholich, K. Behind the Wall-Compartment-Specific Neovascularisation during Post-Stroke Recovery in Mice. Cells 2022, 11, 1659. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Orabona, C.; Puccetti, P.; Vacca, C.; Bicciato, S.; Luchini, A.; Fallarino, F.; Bianchi, R.; Velardi, E.; Perruccio, K.; Velardi, A.; et al. Toward the identification of a tolerogenic signature in IDO-competent dendritic cells. Blood 2006, 107, 2846–2854. [Google Scholar] [CrossRef]
- Li, H.; Yuan, Y.; Chen, H.; Dai, H.; Li, J. Indoleamine 2,3-dioxygenase mediates the therapeutic effects of adipose-derived stromal/stem cells in experimental periodontitis by modulating macrophages through the kynurenine-AhR-NRF2 pathway. Mol. Metab. 2022, 66, 101617. [Google Scholar] [CrossRef]
- Tacke, F.; Puengel, T.; Loomba, R.; Friedman, S.L. An integrated view of anti-inflammatory and antifibrotic targets for the treatment of NASH. J. Hepatol. 2023, in press. [Google Scholar] [CrossRef]
- Engelmann, C.; Tacke, F. The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 652. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Che, D.; Qin, G.; Farouk, M.H.; Hailong, J.; Rui, H. Novel Biosynthesis, Metabolism and Physiological Functions of L-Homoarginine. Curr. Protein Pept. Sci. 2019, 20, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Jia, S.; Nawaratna, G.; Hu, S.; Dahanayaka, S.; Bazer, F.W.; Wu, G. Analysis of L-homoarginine in biological samples by HPLC involving precolumn derivatization with o-phthalaldehyde and N-acetyl-L-cysteine. Amino Acids 2015, 47, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- März, W.; Meinitzer, A.; Drechsler, C.; Pilz, S.; Krane, V.; Kleber, M.E.; Fischer, J.; Winkelmann, B.R.; Böhm, B.O.; Ritz, E.; et al. Homoarginine, cardiovascular risk, and mortality. Circulation 2010, 122, 967–975. [Google Scholar] [CrossRef]
- Koch, V.; Gruenewald, L.D.; Gruber-Rouh, T.; Eichler, K.; Leistner, D.M.; Mahmoudi, S.; Booz, C.; Bernatz, S.; D’Angelo, T.; Albrecht, M.H.; et al. Homoarginine in the cardiovascular system: Pathophysiology and recent developments. Fundam. Clin. Pharmacol. 2022, 37, 519–529. [Google Scholar] [CrossRef]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef]
- Murray, M.F. Tryptophan depletion and HIV infection: A metabolic link to pathogenesis. Lancet. Infect. Dis. 2003, 3, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Scholich, K. Toponomics: Studying protein-protein interactions and protein networks in intact tissue. Mol. Biosyst. 2010, 6, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Linke, B.; Suo, J.; Tarighi, N.; Del Turco, D.; Thomas, D.; Ferreiros, N.; Stegner, D.; Frolich, S.; Sisignano, M.; et al. GPVI and Thromboxane Receptor on Platelets Promote Proinflammatory Macrophage Phenotypes during Cutaneous Inflammation. J. Investig. Dermatol. 2017, 137, 686–695. [Google Scholar] [CrossRef]
- Kornstädt, L.; Pierre, S.; Weigert, A.; Ebersberger, S.; Schäufele, T.J.; Kolbinger, A.; Schmid, T.; Cohnen, J.; Thomas, D.; Ferreirós, N.; et al. Bacterial and Fungal Toll-Like Receptor Activation Elicits Type I IFN Responses in Mast Cells. Front. Immunol. 2020, 11, 607048. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Varela, M.; Paish, H.; Alvarez, L.; Uriarte, I.; Latasa, M.U.; Santamaria, E.; Recalde, M.; Garate, M.; Claveria, A.; Colyn, L.; et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 2021, 70, 388–400. [Google Scholar] [CrossRef]
- Yarnell, A.M.; Barry, E.S.; Mountney, A.; Shear, D.; Tortella, F.; Grunberg, N.E. The Revised Neurobehavioral Severity Scale (NSS-R) for Rodents. Curr. Protoc. Neurosci. 2016, 75, e104. [Google Scholar] [CrossRef]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef]
- Dorochow, E.; Gurke, R.; Rischke, S.; Geisslinger, G.; Hahnefeld, L. Effects of Different Storage Conditions on Lipid Stability in Mice Tissue Homogenates. Metabolites 2023, 13, 504. [Google Scholar] [CrossRef]
- Sens, A.; Rischke, S.; Hahnefeld, L.; Dorochow, E.; Schäfer, S.M.G.; Thomas, D.; Köhm, M.; Geisslinger, G.; Behrens, F.; Gurke, R. Pre-analytical sample handling standardization for reliable measurement of metabolites and lipids in LC-MS-based clinical research. J. Mass Spectrom. Adv. Clin. Lab. 2023, 28, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Kratz, D.; Wilken-Schmitz, A.; Sens, A.; Hahnefeld, L.; Scholich, K.; Geisslinger, G.; Gurke, R.; Thomas, D. Post-mortem changes of prostanoid concentrations in tissues of mice: Impact of fast cervical dislocation and dissection delay. Prostaglandins Other Lipid Mediat. 2022, 162, 106660. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorochow, E.; Kraus, N.; Chenaux-Repond, N.; Pierre, S.; Kolbinger, A.; Geisslinger, G.; Ortiz, C.; Welsch, C.; Trebicka, J.; Gurke, R.; et al. Differential Lipidomics, Metabolomics and Immunological Analysis of Alcoholic and Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2023, 24, 10351. https://doi.org/10.3390/ijms241210351
Dorochow E, Kraus N, Chenaux-Repond N, Pierre S, Kolbinger A, Geisslinger G, Ortiz C, Welsch C, Trebicka J, Gurke R, et al. Differential Lipidomics, Metabolomics and Immunological Analysis of Alcoholic and Non-Alcoholic Steatohepatitis in Mice. International Journal of Molecular Sciences. 2023; 24(12):10351. https://doi.org/10.3390/ijms241210351
Chicago/Turabian StyleDorochow, Erika, Nico Kraus, Nicolas Chenaux-Repond, Sandra Pierre, Anja Kolbinger, Gerd Geisslinger, Cristina Ortiz, Christoph Welsch, Jonel Trebicka, Robert Gurke, and et al. 2023. "Differential Lipidomics, Metabolomics and Immunological Analysis of Alcoholic and Non-Alcoholic Steatohepatitis in Mice" International Journal of Molecular Sciences 24, no. 12: 10351. https://doi.org/10.3390/ijms241210351
APA StyleDorochow, E., Kraus, N., Chenaux-Repond, N., Pierre, S., Kolbinger, A., Geisslinger, G., Ortiz, C., Welsch, C., Trebicka, J., Gurke, R., Hahnefeld, L., Klein, S., & Scholich, K. (2023). Differential Lipidomics, Metabolomics and Immunological Analysis of Alcoholic and Non-Alcoholic Steatohepatitis in Mice. International Journal of Molecular Sciences, 24(12), 10351. https://doi.org/10.3390/ijms241210351