
The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
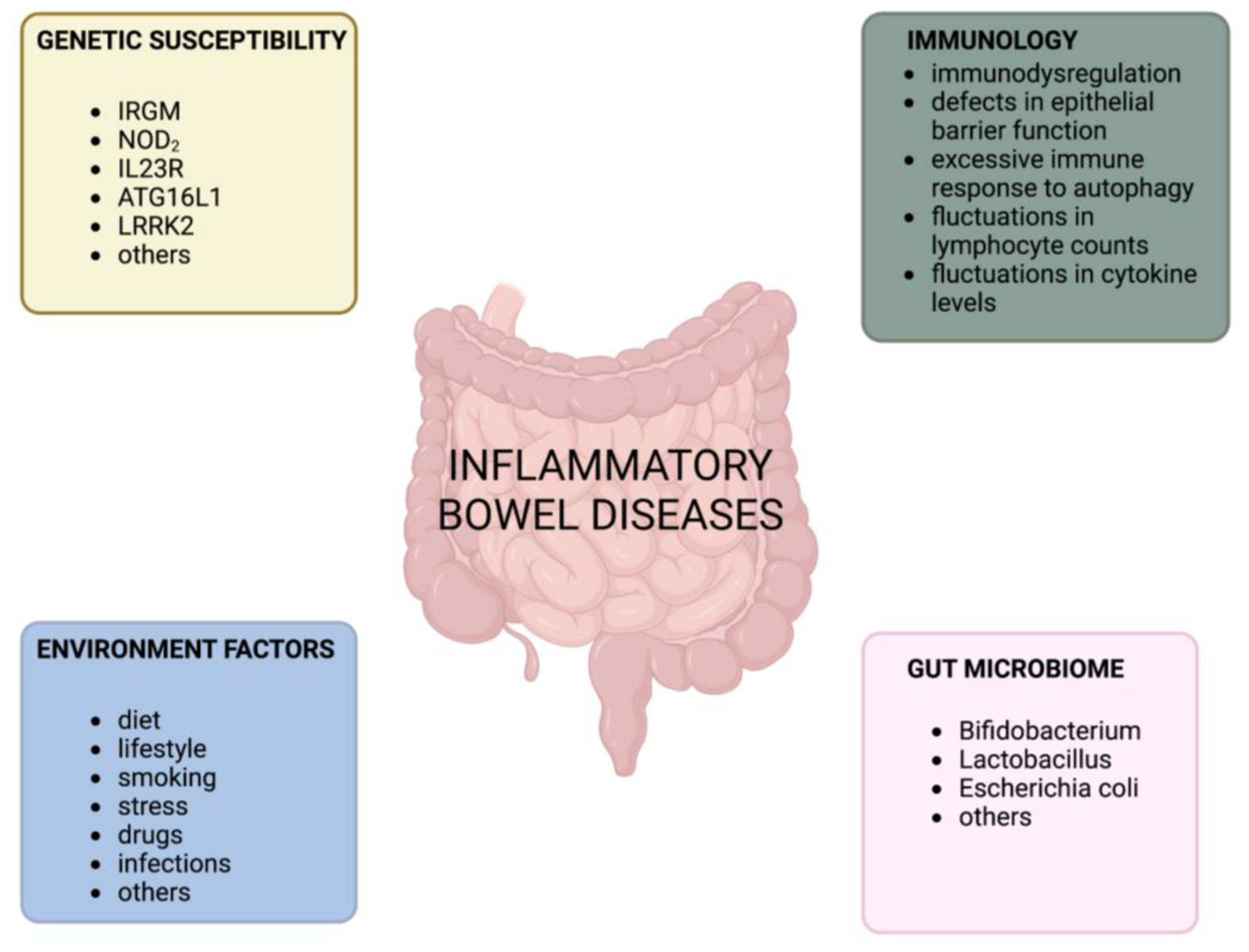
1. Introduction
2. Biological Function of IL-23
3. Factors Controlling the Production of IL-23
3.1. Microbial Stimulation
3.2. IL-1 Cytokine Network
3.3. The cAMP Pathway
3.4. CXCL8 Chemokine Family
4. Effectors of the IL-23 Response
4.1. The IL-23-Th17 Axis
4.2. Innate Lymphoid Cells and the IL-23-IL-22 Axis
4.3. Regulatory T Cells
5. Myeloid Cells
6. IL-23 and IBD
7. Intestinal Homeostasis—Mice Models of Inflammatory Bowel Diseases
8. Genetic Correlations
9. Treatment
9.1. The First Generation
9.2. The Second Generation
9.3. The Third Generation
9.4. Results of Treatment
10. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahluwalia, B.; Magnusson, M.K.; Öhman, L. Mucosal Immune System of the Gastrointestinal Tract: Maintaining Balance between the Good and the Bad. Scand. J. Gastroenterol. 2017, 52, 1185–1193. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Powrie, F. Translating Immunology into Therapeutic Concepts for Inflammatory Bowel Disease. Annu. Rev. Immunol. 2018, 36, 755–781. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Gomollón, F.; Dignass, A.; Annese, V.; Tilg, H.; Van Assche, G.; Lindsay, J.O.; Peyrin-Biroulet, L.; Cullen, G.J.; Daperno, M.; Kucharzik, T.; et al. 3rd European Evidence-Based Consensus on the Diagnosis and Management of Crohn’s Disease 2016: Part 1: Diagnosis and Medical Management. J. Crohns Colitis 2017, 11, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.L. Immunopathology of Inflammatory Bowel Disease. World J. Gastroenterol. 2014, 20, 6. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the Pathogenesis of Inflammatory Bowel Disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-R. Dysregulation of Mucosal Immune Response in Pathogenesis of Inflammatory Bowel Disease. World J. Gastroenterol. 2014, 20, 3255. [Google Scholar] [CrossRef]
- De Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current State of the Art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Swidsinski, A.; Ladhoff, A.; Pernthaler, A.; Swidsinski, S.; Loening–Baucke, V.; Ortner, M.; Weber, J.; Hoffmann, U.; Schreiber, S.; Dietel, M.; et al. Mucosal Flora in Inflammatory Bowel Disease. Gastroenterology 2002, 122, 44–54. [Google Scholar] [CrossRef]
- Zhang, Y.-Z. Inflammatory Bowel Disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91. [Google Scholar] [CrossRef]
- Schirbel, A.; Fiocchi, C. Inflammatory Bowel Disease: Established and Evolving Considerations on Its Etiopathogenesis and Therapy: IBD Etiopathogenesis and Therapy. J. Dig. Dis. 2010, 11, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Ha, E.; Gu, K.-N.; Shin, G.Y.; Lee, C.K.; Kim, K.; Kim, H.J. Host Genetic and Gut Microbial Signatures in Familial Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2020, 11, e00213. [Google Scholar] [CrossRef] [PubMed]
- The International IBD Genetics Consortium (IIBDGC); Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; et al. Host–Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance Infliximab for Crohn’s Disease: The ACCENT I Randomised Trial. Lancet 2002, 359, 1541–1549. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Van Assche, G.; Reinisch, W.; Colombel, J.; D’Haens, G.; Wolf, D.C.; Kron, M.; Tighe, M.B.; Lazar, A.; Thakkar, R.B. Adalimumab Induces and Maintains Clinical Remission in Patients with Moderate-to-Severe Ulcerative Colitis. Gastroenterology 2012, 142, 257–265.e3. [Google Scholar] [CrossRef]
- Peña-Sánchez, J.N.; Osei, J.A.; Marques Santos, J.D.; Jennings, D.; Andkhoie, M.; Brass, C.; Bukassa-Kazadi, G.; Lu, X.; Johnson-Jennings, M.; Porter, L.; et al. Increasing Prevalence and Stable Incidence Rates of Inflammatory Bowel Disease Among First Nations: Population-Based Evidence From a Western Canadian Province. Inflamm. Bowel Dis. 2022, 28, 514–522. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The Inhibitory Cytokine IL-35 Contributes to Regulatory T-Cell Function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Wang, R.-X.; Yu, C.-R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.-H.; Egwuagu, C.E. Interleukin-35 Induces Regulatory B Cells That Suppress Autoimmune Disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef]
- Hunter, C.A. New IL-12-Family Members: IL-23 and IL-27, Cytokines with Divergent Functions. Nat. Rev. Immunol. 2005, 5, 521–531. [Google Scholar] [CrossRef]
- Trinchieri, G.; Pflanz, S.; Kastelein, R.A. The IL-12 Family of Heterodimeric Cytokines. Immunity 2003, 19, 641–644. [Google Scholar] [CrossRef]
- Maloy, K.J.; Kullberg, M.C. IL-23 and Th17 Cytokines in Intestinal Homeostasis. Mucosal Immunol. 2008, 1, 339–349. [Google Scholar] [CrossRef]
- Neurath, M.F. IL-23 in Inflammatory Bowel Diseases and Colon Cancer. Cytokine Growth Factor Rev. 2019, 45, 1–8. [Google Scholar] [CrossRef]
- Jefremow, A.; Neurath, M.F. All Are Equal, Some Are More Equal: Targeting IL 12 and 23 in IBD—A Clinical Perspective. Immuno Targets Ther. 2020, 9, 289–297. [Google Scholar] [CrossRef]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and Biology of IL-23 and IL-27: Related but Functionally Distinct Regulators of Inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef]
- Yoshiga, Y.; Goto, D.; Segawa, S.; Ohnishi, Y.; Matsumoto, I.; Ito, S.; Tsutsumi, A.; Taniguchi, M.; Sumida, T. Invariant NKT Cells Produce IL-17 through IL-23-Dependent and -Independent Pathways with Potential Modulation of Th17 Response in Collagen-Induced Arthritis. Int. J. Mol. Med. 2008, 22, 369–374. [Google Scholar] [CrossRef]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel P19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Toy, T.; Shin, A.; Wagner, M.; Cebon, J.; Maraskovsky, E. Extracellular Nucleotide Signaling by P2 Receptors Inhibits IL-12 and Enhances IL-23 Expression in Human Dendritic Cells: A Novel Role for the CAMP Pathway. Blood 2005, 105, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. J. Crohns Colitis 2022, 16 (Suppl. 2), ii3–ii19. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Chinen, H.; Kobayashi, T.; Sato, T.; Sakuraba, A.; Kitazume, M.T.; Sugita, A.; Koganei, K.; et al. Unique CD14+ Intestinal Macrophages Contribute to the Pathogenesis of Crohn Disease via IL-23/IFN-γ Axis. J. Clin. Investig. 2008, 118, 2269–2280. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A Microbial Signature for Crohn’s Disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Liu, Z.; Yadav, P.K.; Xu, X.; Su, J.; Chen, C.; Tang, M.; Lin, H.; Yu, J.; Qian, J.; Yang, P.-C.; et al. The Increased Expression of IL-23 in Inflammatory Bowel Disease Promotes Intraepithelial and Lamina Propria Lymphocyte Inflammatory Responses and Cytotoxicity. J. Leukoc. Biol. 2011, 89, 597–606. [Google Scholar] [CrossRef]
- Becker, C.; Wirtz, S.; Blessing, M.; Pirhonen, J.; Strand, D.; Bechthold, O.; Frick, J.; Galle, P.R.; Autenrieth, I.; Neurath, M.F. Constitutive P40 Promoter Activation and IL-23 Production in the Terminal Ileum Mediated by Dendritic Cells. J. Clin. Investig. 2003, 112, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Brain, O.; Owens, B.M.J.; Pichulik, T.; Allan, P.; Khatamzas, E.; Leslie, A.; Steevels, T.; Sharma, S.; Mayer, A.; Catuneanu, A.M.; et al. The Intracellular Sensor NOD2 Induces MicroRNA-29 Expression in Human Dendritic Cells to Limit IL-23 Release. Immunity 2013, 39, 521–536. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic Regulation of the Immune System. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Haas, J.T.; L’homme, L.; Fleury, S.; Quemener, S.; Levavasseur, M.; Becquart, C.; Wartelle, J.; Bogomolova, A.; Pineau, L.; et al. Metabolic and Innate Immune Cues Merge into a Specific Inflammatory Response via the UPR. Cell 2019, 177, 1201–1216.e19. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Boroviak, K.; Zhang, Q.; Assadi, G.; Kempster, S.L.; Sewell, G.W.; Saveljeva, S.; Ashcroft, J.W.; Clare, S.; Mukhopadhyay, S.; et al. C13orf31 (FAMIN) Is a Central Regulator of Immunometabolic Function. Nat. Immunol. 2016, 17, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.P.; Markhard, A.L.; Shah, H.; Sharma, R.; Skinner, O.S.; Clish, C.B.; Deik, A.; Patgiri, A.; Hsu, Y.-H.H.; Masia, R.; et al. Hepatic NADH Reductive Stress Underlies Common Variation in Metabolic Traits. Nature 2020, 583, 122–126. [Google Scholar] [CrossRef]
- Cader, M.Z.; De Almeida Rodrigues, R.P.; West, J.A.; Sewell, G.W.; Md-Ibrahim, M.N.; Reikine, S.; Sirago, G.; Unger, L.W.; Iglesias-Romero, A.B.; Ramshorn, K.; et al. FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell 2020, 180, 278–295.e23. [Google Scholar] [CrossRef]
- Aschenbrenner, D.; Quaranta, M.; Banerjee, S.; Ilott, N.; Jansen, J.; Steere, B.; Chen, Y.-H.; Ho, S.; Cox, K.; Arancibia-Cárcamo, C.V.; et al. Deconvolution of Monocyte Responses in Inflammatory Bowel Disease Reveals an IL-1 Cytokine Network That Regulates IL-23 in Genetic and Acquired IL-10 Resistance. Gut 2021, 70, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Pflanz, S.; Timans, J.C.; Cheung, J.; Rosales, R.; Kanzler, H.; Gilbert, J.; Hibbert, L.; Churakova, T.; Travis, M.; Vaisberg, E.; et al. IL-27, a Heterodimeric Cytokine Composed of EBI3 and P28 Protein, Induces Proliferation of Naive CD4+ T Cells. Immunity 2002, 16, 779–790. [Google Scholar] [CrossRef]
- Veckman, V.; Miettinen, M.; Pirhonen, J.; Sirén, J.; Matikainen, S.; Julkunen, I. Streptococcus Pyogenes and Lactobacillus Rhamnosus Differentially Induce Maturation and Production of Th1-Type Cytokines and Chemokines in Human Monocyte-Derived Dendritic Cells. J. Leukoc. Biol. 2004, 75, 764–771. [Google Scholar] [CrossRef]
- Van Seventer, J.M.; Nagai, T.; Van Seventer, G.A. Interferon-β Differentially Regulates Expression of the IL-12 Family Members P35, P40, P19 and EBI3 in Activated Human Dendritic Cells. J. Neuroimmunol. 2002, 133, 60–71. [Google Scholar] [CrossRef]
- Wesa, A.; Galy, A. Increased Production of Pro-Inflammatory Cytokines and Enhanced T Cell Responses after Activation of Human Dendritic Cells with IL-1 and CD40 Ligand. BMC Immunol. 2002, 3, 14. [Google Scholar] [CrossRef]
- Sakuraba, A.; Sato, T.; Kamada, N.; Kitazume, M.; Sugita, A.; Hibi, T. Th1/Th17 Immune Response Is Induced by Mesenteric Lymph Node Dendritic Cells in Crohn’s Disease. Gastroenterology 2009, 137, 1736–1745. [Google Scholar] [CrossRef]
- Verreck, F.A.W.; De Boer, T.; Langenberg, D.M.L.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; De Waal-Malefyt, R.; Ottenhoff, T.H.M. Human IL-23-Producing Type 1 Macrophages Promote but IL-10-Producing Type 2 Macrophages Subvert Immunity to (Myco)Bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef]
- Snijders, A. High-Level IL-12 Production by Human Dendritic Cells Requires Two Signals. Int. Immunol. 1998, 10, 1593–1598. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Lourda, M.; Ideström, M.; Chen, P.; Olsson-Åkefeldt, S.; Forkel, M.; Gavhed, D.; Lindforss, U.; Mjösberg, J.; Henter, J.-I.; et al. Tissue-Infiltrating Neutrophils Represent the Main Source of IL-23 in the Colon of Patients with IBD. Gut 2016, 65, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 Chemokine Family and Its Receptors in Inflammatory Diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming Growth Factor-β Induces Development of the TH17 Lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Liang, S.C.; Tan, X.-Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 Are Coexpressed by Th17 Cells and Cooperatively Enhance Expression of Antimicrobial Peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Bending, D.; De La Peña, H.; Veldhoen, M.; Phillips, J.M.; Uyttenhove, C.; Stockinger, B.; Cooke, A. Highly Purified Th17 Cells from BDC2.5NOD Mice Convert into Th1-like Cells in NOD/SCID Recipient Mice. J. Clin. Investig. 2009, 119, 565–572. [Google Scholar] [CrossRef]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C.; Tolaini, M.; Menzel, U.; et al. Fate Mapping of IL-17-Producing T Cells in Inflammatory Responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Billmeier, U.; Dieterich, W.; Rath, T.; Sonnewald, S.; Reid, S.; Hirschmann, S.; Hildner, K.; Waldner, M.J.; Mudter, J.; et al. Expansion of IL-23 Receptor Bearing TNFR2+ T Cells Is Associated with Molecular Resistance to Anti-TNF Therapy in Crohn’s Disease. Gut 2019, 68, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Globig, A.-M.; Hennecke, N.; Martin, B.; Seidl, M.; Ruf, G.; Hasselblatt, P.; Thimme, R.; Bengsch, B. Comprehensive Intestinal T Helper Cell Profiling Reveals Specific Accumulation of IFN-Γ+IL-17+Coproducing CD4+ T Cells in Active Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2014, 20, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Kamada, N.; Chinen, H.; Okamoto, S.; Kitazume, M.T.; Chang, J.; Matuzaki, Y.; Suzuki, S.; Sugita, A.; Koganei, K.; et al. Imbalance of NKp44+NKp46− and NKp44−NKp46+ Natural Killer Cells in the Intestinal Mucosa of Patients With Crohn’s Disease. Gastroenterology 2010, 139, 882–892.e3. [Google Scholar] [CrossRef] [PubMed]
- Almradi, A.; Hanzel, J.; Sedano, R.; Parker, C.E.; Feagan, B.G.; Ma, C.; Jairath, V. Clinical Trials of IL-12/IL-23 Inhibitors in Inflammatory Bowel Disease. BioDrugs 2020, 34, 713–721. [Google Scholar] [CrossRef]
- Noviello, D.; Mager, R.; Roda, G.; Borroni, R.G.; Fiorino, G.; Vetrano, S. The IL23-IL17 Immune Axis in the Treatment of Ulcerative Colitis: Successes, Defeats, and Ongoing Challenges. Front. Immunol. 2021, 12, 611256. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.-P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of Pathogenic TH17 Cells in the Absence of TGF-β Signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and Therapeutic Targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 Cytokines Stimulate CCL20 Expression in Keratinocytes In Vitro and In Vivo: Implications for Psoriasis Pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 Immune Axis: From Mechanisms to Therapeutic Testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Chen, F.; Cao, A.; Yao, S.; Evans-Marin, H.L.; Liu, H.; Wu, W.; Carlsen, E.D.; Dann, S.M.; Soong, L.; Sun, J.; et al. MTOR Mediates IL-23 Induction of Neutrophil IL-17 and IL-22 Production. J. Immunol. 2016, 196, 4390–4399. [Google Scholar] [CrossRef]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic Bacteria Direct Expression of an Intestinal Bactericidal Lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef]
- Kinugasa, T.; Sakaguchi, T.; Gu, X.; Reinecker, H. Claudins Regulate the Intestinal Barrier in Response to Immune Mediators. Gastroenterology 2000, 118, 1001–1011. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Cader, M.Z.; Kaser, A. Recent Advances in Inflammatory Bowel Disease: Mucosal Immune Cells in Intestinal Inflammation. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.Y.; Bealgey, K.W.; Fang, Y.; Gong, Y.M.; Bao, S. Interleukin-23: Immunological Roles and Clinical Implications. Int. J. Biochem. Cell Biol. 2009, 41, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.-L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 Cytokines: From Discovery to Targeted Therapies for Immune-Mediated Inflammatory Diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Takatori, H.; Kanno, Y.; Watford, W.T.; Tato, C.M.; Weiss, G.; Ivanov, I.I.; Littman, D.R.; O’Shea, J.J. Lymphoid Tissue Inducer–like Cells Are an Innate Source of IL-17 and IL-22. J. Exp. Med. 2009, 206, 35–41. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.M.; Doherty, J.M.; Mills, J.C.; Colonna, M. A Human Natural Killer Cell Subset Provides an Innate Source of IL-22 for Mucosal Immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX3CR1+ Mononuclear Phagocytes Support Colitis-Associated Innate Lymphoid Cell Production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, J.; Tu, T.; Yang, X.; Deng, L.; Anders, R.A.; Zhou, L.; Fu, Y.-X. Induction of Innate Lymphoid Cell-Derived Interleukin-22 by the Transcription Factor STAT3 Mediates Protection against Intestinal Infection. Immunity 2014, 40, 25–39. [Google Scholar] [CrossRef]
- Bauché, D.; Joyce-Shaikh, B.; Fong, J.; Villarino, A.V.; Ku, K.S.; Jain, R.; Lee, Y.; Annamalai, L.; Yearley, J.H.; Cua, D.J. IL-23 and IL-2 Activation of STAT5 Is Required for Optimal IL-22 Production in ILC3s during Colitis. Sci. Immunol. 2020, 5, eaav1080. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Cárcamo, C.V.; Fleming, M.P.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.L.; Powrie, F. IL-23–Responsive Innate Lymphoid Cells Are Increased in Inflammatory Bowel Disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef]
- Keir, M.E.; Yi, T.; Lu, T.T.; Ghilardi, N. The Role of IL-22 in Intestinal Health and Disease. J. Exp. Med. 2020, 217, e20192195. [Google Scholar] [CrossRef] [PubMed]
- Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, J. Role of Th17 Cells in the Pathogenesis of Human IBD. ISRN Inflamm. 2014, 2014, 928461. [Google Scholar] [CrossRef]
- Ito, Y.; Usui, T.; Kobayashi, S.; Iguchi-Hashimoto, M.; Ito, H.; Yoshitomi, H.; Nakamura, T.; Shimizu, M.; Kawabata, D.; Yukawa, N.; et al. Gamma/Delta T Cells Are the Predominant Source of Interleukin-17 in Affected Joints in Collagen-Induced Arthritis, but Not in Rheumatoid Arthritis. Arthritis Rheum. 2009, 60, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, E.; Green, A.M.; Flynn, J.L. IL-17 Production Is Dominated by Γδ T Cells Rather than CD4 T Cells during Mycobacterium Tuberculosis Infection. J. Immunol. 2006, 177, 4662–4669. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Hue, S.; Buonocore, S.; Arancibia-Cárcamo, C.V.; Ahern, P.P.; Iwakura, Y.; Maloy, K.J.; Powrie, F. Interleukin-23 Restrains Regulatory T Cell Activity to Drive T Cell-Dependent Colitis. Immunity 2008, 28, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 Programs TH-17 Cell Differentiation by Promoting Sequential Engagement of the IL-21 and IL-23 Pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory Lymphocytes and Intestinal Inflammation. Annu. Rev. Immunol. 2009, 27, 313–338. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 Drives Intestinal Inflammation through Direct Activity on T Cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef]
- Bauché, D.; Joyce-Shaikh, B.; Jain, R.; Grein, J.; Ku, K.S.; Blumenschein, W.M.; Ganal-Vonarburg, S.C.; Wilson, D.C.; McClanahan, T.K.; Malefyt, R.D.W.; et al. LAG3+ Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1+ Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity 2018, 49, 342–352.e5. [Google Scholar] [CrossRef]
- Fournier, B.M.; Parkos, C.A. The Role of Neutrophils during Intestinal Inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Lees, C.W.; Barrett, J.C.; Parkes, M.; Satsangi, J. New IBD Genetics: Common Pathways with Other Diseases. Gut 2011, 60, 1739–1753. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and Immunity: Challenges and Opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Griseri, T.; McKenzie, B.S.; Schiering, C.; Powrie, F. Dysregulated Hematopoietic Stem and Progenitor Cell Activity Promotes Interleukin-23-Driven Chronic Intestinal Inflammation. Immunity 2012, 37, 1116–1129. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate Lymphoid Cells Drive Interleukin-23-Dependent Innate Intestinal Pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.J.; Falkowski, N.R.; McDonald, R.A.; Pandit, C.R.; Young, V.B.; Huffnagle, G.B. Interleukin-23 (IL-23), Independent of IL-17 and IL-22, Drives Neutrophil Recruitment and Innate Inflammation during Clostridium difficile Colitis in Mice. Immunology 2016, 147, 114–124. [Google Scholar] [CrossRef]
- Wu, Q.; Martin, R.J.; Rino, J.G.; Breed, R.; Torres, R.M.; Chu, H.W. IL-23-Dependent IL-17 Production Is Essential in Neutrophil Recruitment and Activity in Mouse Lung Defense against Respiratory Mycoplasma Pneumoniae Infection. Microbes Infect. 2007, 9, 78–86. [Google Scholar] [CrossRef]
- Smith, E.; Zarbock, A.; Stark, M.A.; Burcin, T.L.; Bruce, A.C.; Foley, P.; Ley, K. IL-23 Is Required for Neutrophil Homeostasis in Normal and Neutrophilic Mice. J. Immunol. 2007, 179, 8274–8279. [Google Scholar] [CrossRef] [PubMed]
- Griseri, T.; Arnold, I.C.; Pearson, C.; Krausgruber, T.; Schiering, C.; Franchini, F.; Schulthess, J.; McKenzie, B.S.; Crocker, P.R.; Powrie, F. Granulocyte Macrophage Colony-Stimulating Factor-Activated Eosinophils Promote Interleukin-23 Driven Chronic Colitis. Immunity 2015, 43, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Kayama, H.; Tani, H.; Kitada, S.; Opasawatchai, A.; Okumura, R.; Motooka, D.; Nakamura, S.; Takeda, K. BATF2 Prevents T-Cell-Mediated Intestinal Inflammation through Regulation of the IL-23/IL-17 Pathway. Int. Immunol. 2019, 31, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Bernshtein, B.; Curato, C.; Ioannou, M.; Thaiss, C.A.; Gross-Vered, M.; Kolesnikov, M.; Wang, Q.; David, E.; Chappell-Maor, L.; Harmelin, A.; et al. IL-23–Producing IL-10Rα–Deficient Gut Macrophages Elicit an IL-22–Driven Proinflammatory Epithelial Cell Response. Sci. Immunol. 2019, 4, eaau6571. [Google Scholar] [CrossRef]
- Tamassia, N.; Arruda-Silva, F.; Wright, H.L.; Moots, R.J.; Gardiman, E.; Bianchetto-Aguilera, F.; Gasperini, S.; Capone, M.; Maggi, L.; Annunziato, F.; et al. Human Neutrophils Activated via TLR8 Promote Th17 Polarization through IL-23. J. Leukoc. Biol. 2019, 105, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Hedl, M.; Abraham, C. IL23 Induces IL23R Recycling and Amplifies Innate Receptor-Induced Signalling and Cytokines in Human Macrophages, and the IBD-Protective IL23R R381Q Variant Modulates These Outcomes. Gut 2020, 69, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.L.; Mair, F.; Becher, B. IL-23: One Cytokine in Control of Autoimmunity: HIGHLIGHTS. Eur. J. Immunol. 2012, 42, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Bouma, G.; Strober, W. The Immunological and Genetic Basis of Inflammatory Bowel Disease. Nat. Rev. Immunol. 2003, 3, 521–533. [Google Scholar] [CrossRef]
- Lim, K.S.; Yong, Z.W.E.; Wang, H.; Tan, T.Z.; Huang, R.Y.-J.; Yamamoto, D.; Inaki, N.; Hazawa, M.; Wong, R.W.; Oshima, H.; et al. Inflammatory and Mitogenic Signals Drive Interleukin 23 Subunit Alpha (IL23A) Secretion Independent of IL12B in Intestinal Epithelial Cells. J. Biol. Chem. 2020, 295, 6387–6400. [Google Scholar] [CrossRef]
- Bosmann, M.; Grailer, J.J.; Russkamp, N.F.; Ruemmler, R.; Zetoune, F.S.; Sarma, J.V.; Ward, P.A. CD11c+ Alveolar Macrophages Are a Source of IL-23 During Lipopolysaccharide-Induced Acute Lung Injury. Shock 2013, 39, 447–452. [Google Scholar] [CrossRef]
- Lucaciu, L.A.; Ilieș, M.; Vesa, Ș.C.; Seicean, R.; Din, S.; Iuga, C.A.; Seicean, A. Serum Interleukin (IL)-23 and IL-17 Profile in Inflammatory Bowel Disease (IBD) Patients Could Differentiate between Severe and Non-Severe Disease. J. Pers. Med. 2021, 11, 1130. [Google Scholar] [CrossRef]
- Mirsattari, D.; Seyyedmajidi, M.; Zojaji, H.; Haghazali, M.; Orimi, P.G.; Shoushtarizadeh, T.; Almasi, S. The Relation between the Level of Interleukin-23 with Duration and Severity of Ulcerative Colitis. Gastroenterol. Hepatol. Bed Bench 2012, 5, 49–53. [Google Scholar]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef]
- Raza, A.; Yousaf, W.; Giannella, R.; Shata, M.T. Th17 Cells: Interactions with Predisposing Factors in the Immunopathogenesis of Inflammatory Bowel Disease. Expert Rev. Clin. Immunol. 2012, 8, 161–168. [Google Scholar] [CrossRef]
- Sobolewska-Włodarczyk, A.; Włodarczyk, M.; Talar, M.; Wiśniewska-Jarosińska, M.; Gąsiorowska, A.; Fichna, J. The Association of the Quality of Sleep with Proinflammatory Cytokine Profile in Inflammatory Bowel Disease Patients. Pharmacol. Rep. PR. 2021, 73, 1660–1669. [Google Scholar] [CrossRef]
- Yang, J.; Xu, L. Elevated IL-23R Expression and Foxp3+Rorgt+ Cells in Intestinal Mucosa During Acute and Chronic Colitis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2785–2792. [Google Scholar] [CrossRef]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; O’Connor, W.; Rongvaux, A.; Van Rooijen, N.; Haberman, A.M.; et al. Control of TH17 Cells Occurs in the Small Intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef]
- Gagliani, N.; Vesely, M.C.A.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; De Zoete, M.R.; Licona-Limón, P.; Paiva, R.S.; Ching, T.; et al. Th17 Cells Transdifferentiate into Regulatory T Cells during Resolution of Inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 Rather than Interleukin-12 Is the Critical Cytokine for Autoimmune Inflammation of the Brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ghilardi, N.; Xie, M.-H.; De Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 Drives a Pathogenic T Cell Population That Induces Autoimmune Inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 Drives Innate and T Cell–Mediated Intestinal Inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; Mckenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 Is Essential for T Cell–Mediated Colitis and Promotes Inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef]
- Karaboga, İ.; Demirtas, S.; Karaca, T. Investigation of the Relationship between the Th17/IL-23 Pathway and Innate-Adaptive Immune System in TNBS-Induced Colitis in Rats. Iran. J. Basic Med. Sci. 2017, 20, 870–879. [Google Scholar] [CrossRef]
- Becker, C.; Dornhoff, H.; Neufert, C.; Fantini, M.C.; Wirtz, S.; Huebner, S.; Nikolaev, A.; Lehr, H.-A.; Murphy, A.J.; Valenzuela, D.M.; et al. Cutting Edge: IL-23 Cross-Regulates IL-12 Production in T Cell-Dependent Experimental Colitis. J. Immunol. 2006, 177, 2760–2764. [Google Scholar] [CrossRef]
- Happel, K.I.; Dubin, P.J.; Zheng, M.; Ghilardi, N.; Lockhart, C.; Quinton, L.J.; Odden, A.R.; Shellito, J.E.; Bagby, G.J.; Nelson, S.; et al. Divergent Roles of IL-23 and IL-12 in Host Defense against Klebsiella Pneumoniae. J. Exp. Med. 2005, 202, 761–769. [Google Scholar] [CrossRef]
- Neurath, M.F.; Fuss, I.; Kelsall, B.L.; Stüber, E.; Strober, W. Antibodies to Interleukin 12 Abrogate Established Experimental Colitis in Mice. J. Exp. Med. 1995, 182, 1281–1290. [Google Scholar] [CrossRef]
- Simpson, S.J.; Shah, S.; Comiskey, M.; de Jong, Y.P.; Wang, B.; Mizoguchi, E.; Bhan, A.K.; Terhorst, C. T Cell-Mediated Pathology in Two Models of Experimental Colitis Depends Predominantly on the Interleukin 12/Signal Transducer and Activator of Transcription (Stat)-4 Pathway, but Is Not Conditional on Interferon Gamma Expression by T Cells. J. Exp. Med. 1998, 187, 1225–1234. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Jankovic, D.; Feng, C.G.; Hue, S.; Gorelick, P.L.; McKenzie, B.S.; Cua, D.J.; Powrie, F.; Cheever, A.W.; Maloy, K.J.; et al. IL-23 Plays a Key Role in Helicobacter Hepaticus-Induced T Cell-Dependent Colitis. J. Exp. Med. 2006, 203, 2485–2494. [Google Scholar] [CrossRef]
- Elson, C.O.; Cong, Y.; McCracken, V.J.; Dimmitt, R.A.; Lorenz, R.G.; Weaver, C.T. Experimental Models of Inflammatory Bowel Disease Reveal Innate, Adaptive, and Regulatory Mechanisms of Host Dialogue with the Microbiota. Immunol. Rev. 2005, 206, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Cotsapas, C.; Voight, B.F.; Rossin, E.; Lage, K.; Neale, B.M.; Wallace, C.; Abecasis, G.R.; Barrett, J.C.; Behrens, T.; Cho, J.; et al. Pervasive Sharing of Genetic Effects in Autoimmune Disease. PLoS Genet. 2011, 7, e1002254. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A Genome-Wide Association Study Identifies IL23R as an Inflammatory Bowel Disease Gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and Pathogenesis of Inflammatory Bowel Disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef]
- Cho, J.H.; Brant, S.R. Recent Insights into the Genetics of Inflammatory Bowel Disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence Variants in the Autophagy Gene IRGM and Multiple Other Replicating Loci Contribute to Crohn’s Disease Susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef] [PubMed]
- Sivanesan, D.; Beauchamp, C.; Quinou, C.; Lee, J.; Lesage, S.; Chemtob, S.; Rioux, J.D.; Michnick, S.W. IL23R (Interleukin 23 Receptor) Variants Protective against Inflammatory Bowel Diseases (IBD) Display Loss of Function Due to Impaired Protein Stability and Intracellular Trafficking. J. Biol. Chem. 2016, 291, 8673–8685. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, M.S.; Cho, J.H.; Rioux, J.D.; McGovern, D.P.B.; Wu, J.; Annese, V.; Achkar, J.-P.; Goyette, P.; Scott, R.; Xu, W.; et al. Ulcerative Colitis-Risk Loci on Chromosomes 1p36 and 12q15 Found by Genome-Wide Association Study. Nat. Genet. 2009, 41, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, M.; Goyette, P.; Boucher, G.; Lo, K.S.; Rivas, M.A.; Stevens, C.; Alikashani, A.; Ladouceur, M.; Ellinghaus, D.; Törkvist, L.; et al. Deep Resequencing of GWAS Loci Identifies Rare Variants in CARD9, IL23R and RNF186 That Are Associated with Ulcerative Colitis. PLoS Genet. 2013, 9, e1003723. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, E.S.; Moon, C.M.; Park, J.J.; Kim, T.I.; Kim, W.H.; Cheon, J.H. Genetic Polymorphisms of IL-23R and IL-17A and Novel Insights into Their Associations with Inflammatory Bowel Disease. Gut 2011, 60, 1527–1536. [Google Scholar] [CrossRef]
- Dubinsky, M.C.; Wang, D.; Picornell, Y.; Wrobel, I.; Katzir, L.; Quiros, A.; Dutridge, D.; Wahbeh, G.; Silber, G.; Bahar, R.; et al. IL-23 Receptor (IL-23R) Gene Protects against Pediatric Crohn’s Disease. Inflamm. Bowel Dis. 2007, 13, 511–515. [Google Scholar] [CrossRef]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 Promotes Tumour Incidence and Growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef]
- Jürgens, M.; Laubender, R.P.; Hartl, F.; Weidinger, M.; Seiderer, J.; Wagner, J.; Wetzke, M.; Beigel, F.; Pfennig, S.; Stallhofer, J.; et al. Disease Activity, ANCA, and IL23R Genotype Status Determine Early Response to Infliximab in Patients with Ulcerative Colitis. Am. J. Gastroenterol. 2010, 105, 1811–1819. [Google Scholar] [CrossRef]
- Gheita, T.A.; El Gazzar, I.I.; El-Fishawy, H.S.; Aboul-Ezz, M.A.; Kenawy, S.A. Involvement of IL-23 in Enteropathic Arthritis Patients with Inflammatory Bowel Disease: Preliminary Results. Clin. Rheumatol. 2014, 33, 713–717. [Google Scholar] [CrossRef]
- Aggeletopoulou, I.; Assimakopoulos, S.F.; Konstantakis, C.; Triantos, C. Interleukin 12/Interleukin 23 Pathway: Biological Basis and Therapeutic Effect in Patients with Crohn’s Disease. World J. Gastroenterol. 2018, 24, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.D.; Dyer, E.C.; Rubin, D.T. IL-23 Monoclonal Antibodies for IBD: So Many, So Different? J. Crohns Colitis. 2022, 16 (Suppl. 2), ii42–ii53. [Google Scholar] [CrossRef] [PubMed]
- Deepak, P.; Loftus, E.V. Ustekinumab in Treatment of Crohn’s Disease: Design, Development, and Potential Place in Therapy. Drug Des. Devel. Ther. 2016, 10, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- Engel, T.; Yung, D.E.; Ma, C.; Pariente, B.; WIls, P.; Eliakim, R.; Ungar, B.; Ben-Horin, S.; Kopylov, U. Effectiveness and Safety of Ustekinumab for Crohn’s Disease; Systematic Review and Pooled Analysis of Real-World Evidence. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver. 2019, 51, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Panaccione, R.; Sandborn, W.J.; Gordon, G.L.; Lee, S.D.; Safdi, A.; Sedghi, S.; Feagan, B.G.; Hanauer, S.; Reinisch, W.; Valentine, J.F.; et al. Briakinumab for Treatment of Crohn’s Disease: Results of a Randomized Trial. Inflamm. Bowel Dis. 2015, 21, 1329–1340. [Google Scholar] [CrossRef]
- Wada, Y.; Cardinale, I.; Khatcherian, A.; Chu, J.; Kantor, A.B.; Gottlieb, A.B.; Tatsuta, N.; Jacobson, E.; Barsoum, J.; Krueger, J.G. Apilimod Inhibits the Production of IL-12 and IL-23 and Reduces Dendritic Cell Infiltration in Psoriasis. PLoS ONE 2012, 7, e35069. [Google Scholar] [CrossRef]
- Carmody, R.J.; Ruan, Q.; Liou, H.-C.; Chen, Y.H. Essential Roles of C-Rel in TLR-Induced IL-23 P19 Gene Expression in Dendritic Cells. J. Immunol. 2007, 178, 186–191. [Google Scholar] [CrossRef]
- Grumont, R.; Hochrein, H.; O’Keeffe, M.; Gugasyan, R.; White, C.; Caminschi, I.; Cook, W.; Gerondakis, S. C-Rel Regulates Interleukin 12 P70 Expression in CD8(+) Dendritic Cells by Specifically Inducing P35 Gene Transcription. J. Exp. Med. 2001, 194, 1021–1032. [Google Scholar] [CrossRef]
- Mason, N.; Aliberti, J.; Caamano, J.C.; Liou, H.-C.; Hunter, C.A. Cutting Edge: Identification of c-Rel-Dependent and -Independent Pathways of IL-12 Production during Infectious and Inflammatory Stimuli. J. Immunol. 2002, 168, 2590–2594. [Google Scholar] [CrossRef]
- Sanjabi, S.; Hoffmann, A.; Liou, H.C.; Baltimore, D.; Smale, S.T. Selective Requirement for C-Rel during IL-12 P40 Gene Induction in Macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 12705–12710. [Google Scholar] [CrossRef]
- Sands, B.E.; Chen, J.; Feagan, B.G.; Penney, M.; Rees, W.A.; Danese, S.; Higgins, P.D.R.; Newbold, P.; Faggioni, R.; Patra, K.; et al. Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology 2017, 153, 77–86.e6. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; D’Haens, G.; Panés, J.; Kaser, A.; Ferrante, M.; Louis, E.; Franchimont, D.; Dewit, O.; Seidler, U.; et al. Induction Therapy with the Selective Interleukin-23 Inhibitor Risankizumab in Patients with Moderate-to-Severe Crohn’s Disease: A Randomised, Double-Blind, Placebo-Controlled Phase 2 Study. Lancet Lond. Engl. 2017, 389, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ferrante, M.; Bhandari, B.R.; D’Haens, G.R.; Berliba, E.; Feagan, B.G.; Laskowski, J.; Friedrich, S.; Durante, M.; Tuttle, J. 882—Efficacy and Safety of Anti-Interleukin-23 Therapy with Mirikizumab (LY3074828) in Patients with Moderate-To-Severe Ulcerative Colitis in a Phase 2 Study. Gastroenterology 2018, 154, S1360–S1361. [Google Scholar] [CrossRef]
- D’Haens, G.G.R.; Sandborn, W.J.; Ferrante, M.; Bhandari, B.R.; Berliba, E.; Hibi, T.; Tuttle, J.; Canavan, J.B.; Friedrich, S.; Durante, M.; et al. OP38 Maintenance Treatment with Mirikizumab, a P19-Directed IL-23 Antibody: 52-Week Results in Patients with Moderately-to-Severely Active Ulcerative Colitis. J. Crohns Colitis. 2019, 13 (Suppl. 1), S026–S027. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Peyrin-Biroulet, L.; Higgins, P.D.; Hirai, F.; Belin, R.; Valderas, E.G.; Miller, D.; Morgan-Cox, M.; Naegeli, A.N.; et al. 1003-Efficacy and Safety of Mirikizumab (LY3074828) in a Phase 2 Study of Patients with Crohn’s Disease. Gastroenterology 2019, 156, S-216. [Google Scholar] [CrossRef]
- Baker, K.F.; Isaacs, J.D. Novel Therapies for Immune-Mediated Inflammatory Diseases: What Can We Learn from Their Use in Rheumatoid Arthritis, Spondyloarthritis, Systemic Lupus Erythematosus, Psoriasis, Crohn’s Disease and Ulcerative Colitis? Ann. Rheum. Dis. 2018, 77, 175–187. [Google Scholar] [CrossRef]
- Soendergaard, C.; Bergenheim, F.H.; Bjerrum, J.T.; Nielsen, O.H. Targeting JAK-STAT Signal Transduction in IBD. Pharmacol. Ther. 2018, 192, 100–111. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W.; Study A3921063 Investigators. Tofacitinib, an Oral Janus Kinase Inhibitor, in Active Ulcerative Colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Su, C.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; Niezychowski, W.; et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2017, 376, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Panés, J.; D’Haens, G.R.; Sands, B.E.; Su, C.; Moscariello, M.; Jones, T.; Pedersen, R.; Friedman, G.S.; Lawendy, N.; et al. Safety of Tofacitinib for Treatment of Ulcerative Colitis, Based on 4.4 Years of Data From Global Clinical Trials. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 1541–1550. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Wang, W.; Niezychowski, W.; Study A3921043 Investigators. A Phase 2 Study of Tofacitinib, an Oral Janus Kinase Inhibitor, in Patients with Crohn’s Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2014, 12, 1485–1493.e2. [Google Scholar] [CrossRef] [PubMed]
- Panés, J.; Sandborn, W.J.; Schreiber, S.; Sands, B.E.; Vermeire, S.; D’Haens, G.; Panaccione, R.; Higgins, P.D.R.; Colombel, J.-F.; Feagan, B.G.; et al. Tofacitinib for Induction and Maintenance Therapy of Crohn’s Disease: Results of Two Phase IIb Randomised Placebo-Controlled Trials. Gut 2017, 66, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Danese, S.; Loftus, E.V.; Vermeire, S.; Schreiber, S.; Ritter, T.; Fogel, R.; Mehta, R.; Nijhawan, S.; Kempiński, R.; et al. Filgotinib as Induction and Maintenance Therapy for Ulcerative Colitis (SELECTION): A Phase 2b/3 Double-Blind, Randomised, Placebo-Controlled Trial. Lancet Lond. Engl. 2021, 397, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sanchez Gonzalez, Y.; Zhou, W.; Clark, R.; Xie, W.; Louis, E.; Loftus, E.V.; Panes, J.; Danese, S. Upadacitinib Treatment Improves Symptoms of Bowel Urgency and Abdominal Pain, and Correlates With Quality of Life Improvements in Patients With Moderate to Severe Ulcerative Colitis. J. Crohns Colitis. 2021, 15, 2022–2030. [Google Scholar] [CrossRef]
- Danese, S.; Vermeire, S.; Zhou, W.; Pangan, A.L.; Siffledeen, J.; Greenbloom, S.; Hébuterne, X.; D’Haens, G.; Nakase, H.; Panés, J.; et al. Upadacitinib as Induction and Maintenance Therapy for Moderately to Severely Active Ulcerative Colitis: Results from Three Phase 3, Multicentre, Double-Blind, Randomised Trials. Lancet 2022, 399, 2113–2128. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb. A Phase 2 Randomized, Double-Blind, Placebo-Controlled Study of the Safety and Efficacy of BMS-986165 in Subjects with Moderate to Severe Ulcerative Colitis; Clinical Trial Registration NCT03934216; Clinicaltrials.Gov. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT03934216 (accessed on 20 April 2023).
- Bristol-Myers Squibb. A Phase 2 Randomized, Double-Blind, Placebo-Controlled Study of the Safety, Efficacy, and Biomarker Response of BMS-986165 in Subjects with Moderate to Severe Ulcerative Colitis; Clinical Trial Registration NCT04613518; Clinicaltrials.Gov. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04613518 (accessed on 20 April 2023).
- Bristol-Myers Squibb. A Phase 2 Randomized, Double-Blind, Placebo-Controlled Study of the Safety and Efficacy of BMS-986165 in Subjects with Moderate to Severe Crohn’s Disease; Clinical Trial Registration NCT03599622; Clinicaltrials.Gov. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT03599622 (accessed on 20 April 2023).
- Sandborn, W.J.; Rutgeerts, P.; Gasink, C.; Jacobstein, D.; Zou, B.; Johanns, J.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Ghosh, S.; et al. Long-Term Efficacy and Safety of Ustekinumab for Crohn’s Disease through the Second Year of Therapy. Aliment. Pharmacol. Ther. 2018, 48, 65–77. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef]
- Sands, B.E.; Irving, P.M.; Hoops, T.; Izanec, J.L.; Gao, L.-L.; Gasink, C.; Greenspan, A.; Allez, M.; Danese, S.; Hanauer, S.B.; et al. 775d Ustekinumab Versus Adalimumab for Induction and Maintenance Therapy in Moderate-to-Severe Crohn’s Disease: The SEAVUE Study. Gastroenterology 2021, 161, e30–e31. [Google Scholar] [CrossRef]
- Cheng, X.; Lee, T.-Y.; Ledet, G.; Zemade, G.; Tovera, J.; Campbell, R.; Purro, N.; Annamalai, T.; Masjedizadeh, M.; Liu, D.; et al. 751 Safety, Tolerability, and Pharmacokinetics of PTG-200, an Oral GI-Restricted Peptide Antagonist of IL-23 Receptor, in Normal Healthy Volunteers. Am. J. Gastroenterol. 2019, 114, S439–S440. [Google Scholar] [CrossRef]
- Ferrante, M.; Panaccione, R.; Baert, F.; Bossuyt, P.; Colombel, J.-F.; Danese, S.; Dubinsky, M.; Feagan, B.G.; Hisamatsu, T.; Lim, A.; et al. Risankizumab as Maintenance Therapy for Moderately to Severely Active Crohn’s Disease: Results from the Multicentre, Randomised, Double-Blind, Placebo-Controlled, Withdrawal Phase 3 FORTIFY Maintenance Trial. Lancet 2022, 399, 2031–2046. [Google Scholar] [CrossRef]
- Dubinsky, M.C.; Clemow, D.B.; Hunter Gibble, T.; Li, X.; Vermeire, S.; Hisamatsu, T.; Travis, S.P.L. Clinical Effect of Mirikizumab Treatment on Bowel Urgency in Patients with Moderately to Severely Active Ulcerative Colitis and the Clinical Relevance of Bowel Urgency Improvement for Disease Remission. Crohn’s Colitis 360 2023, 5, otac044. [Google Scholar] [CrossRef] [PubMed]
- Dubinsky, M.C.; Jairath, V.; Feagan, B.G.; Naegeli, A.N.; Tuttle, J.; Morris, N.; Shan, M.; Arora, V.; Lissoos, T.; Agada, N.; et al. Changes in Health-Related Quality of Life and Associations with Improvements in Clinical Efficacy: A Phase 2 Study of Mirikizumab in Patients with Ulcerative Colitis. BMJ Open Gastroenterol. 2023, 10, e001115. [Google Scholar] [CrossRef] [PubMed]
- Parigi, T.L.; Iacucci, M.; Ghosh, S. Blockade of IL-23: What Is in the Pipeline? J. Crohn’s Colitis 2022, 16 (Suppl. 2), ii64–ii72. [Google Scholar] [CrossRef] [PubMed]
- Dudek, P.; Fabisiak, A.; Zatorski, H.; Malecka-Wojciesko, E.; Talar-Wojnarowska, R. Efficacy, Safety and Future Perspectives of JAK Inhibitors in the IBD Treatment. JCM 2021, 10, 5660. [Google Scholar] [CrossRef]
- Rostami-Nejad, M.; Yazdi, M.H.; Nikfar, S.; Rezaie, A.; Abdollahi, M. Potential Vaccines for Treating Crohn’s Disease. Iran. Biomed. J. 2020, 24, 1–14. [Google Scholar] [CrossRef]
- Guan, Q.; Burtnick, H.A.; Qing, G.; Weiss, C.R.; Ma, A.G.; Ma, Y.; Warrington, R.J.; Peng, Z. Employing an IL-23 P19 Vaccine to Block IL-23 Ameliorates Chronic Murine Colitis. Immunotherapy 2013, 5, 1313–1322. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korta, A.; Kula, J.; Gomułka, K. The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 10172. https://doi.org/10.3390/ijms241210172
Korta A, Kula J, Gomułka K. The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2023; 24(12):10172. https://doi.org/10.3390/ijms241210172
Chicago/Turabian StyleKorta, Aleksandra, Julia Kula, and Krzysztof Gomułka. 2023. "The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease" International Journal of Molecular Sciences 24, no. 12: 10172. https://doi.org/10.3390/ijms241210172
APA StyleKorta, A., Kula, J., & Gomułka, K. (2023). The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease. International Journal of Molecular Sciences, 24(12), 10172. https://doi.org/10.3390/ijms241210172