
The Combination of a Human Biomimetic Liver Microphysiology System with BIOLOGXsym, a Quantitative Systems Toxicology (QST) Modeling Platform for Macromolecules, Provides Mechanistic Understanding of Tocilizumab- and GGF2-Induced Liver Injury

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
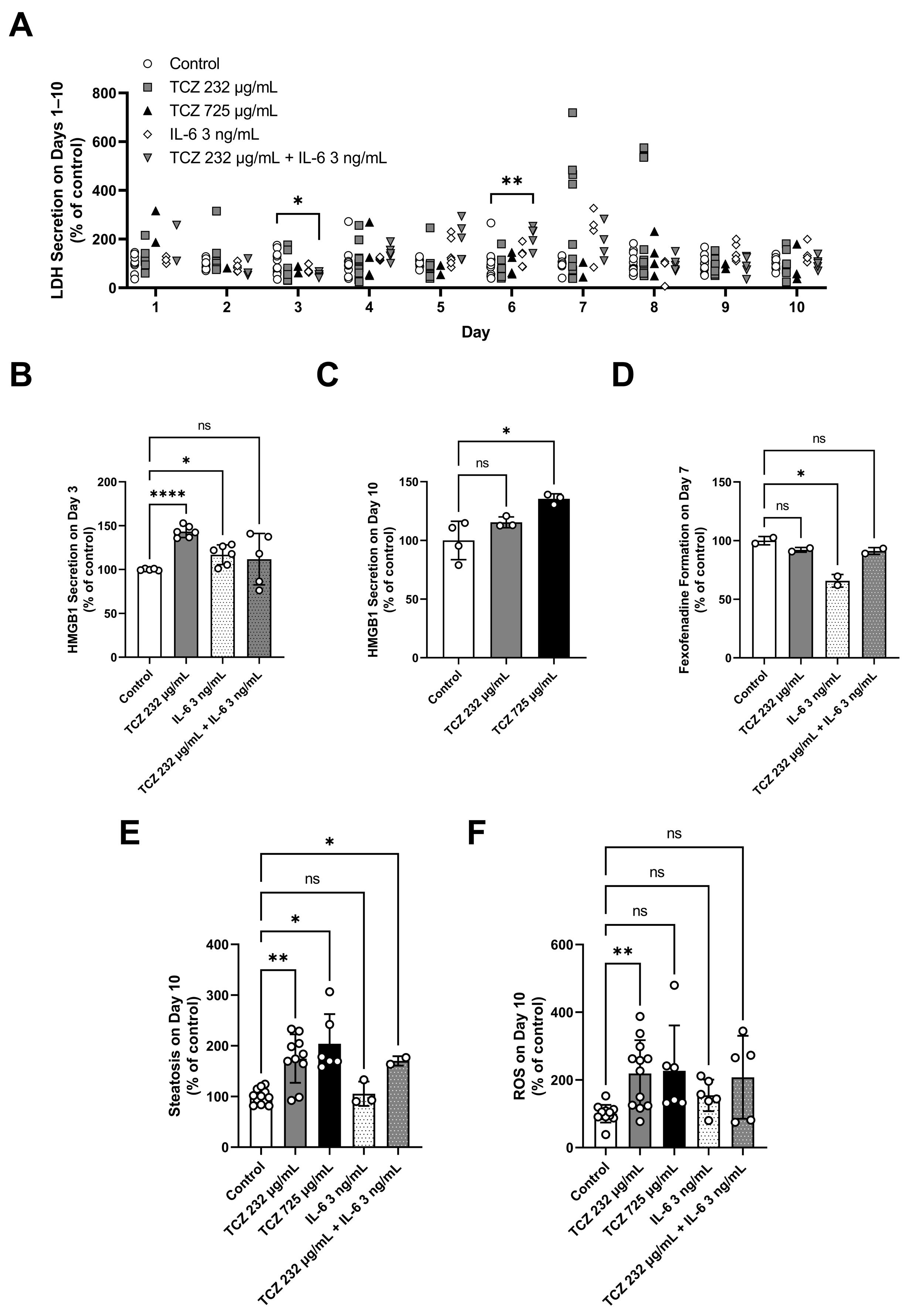
2.1. Evaluation of Tocilizumiab Effects on Phenotypic and Mechanistic Toxicity Endpoints in LAMPS Models
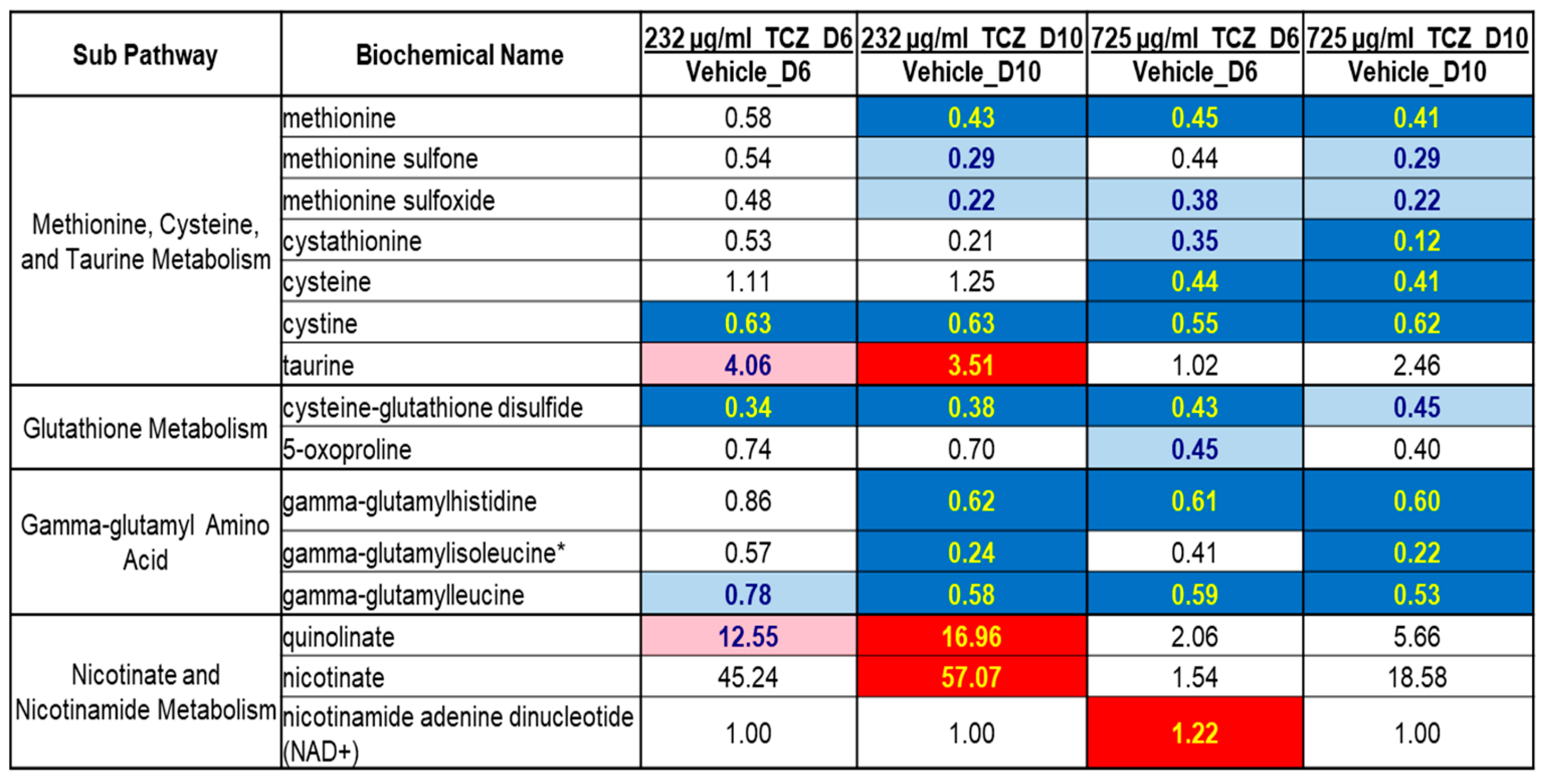
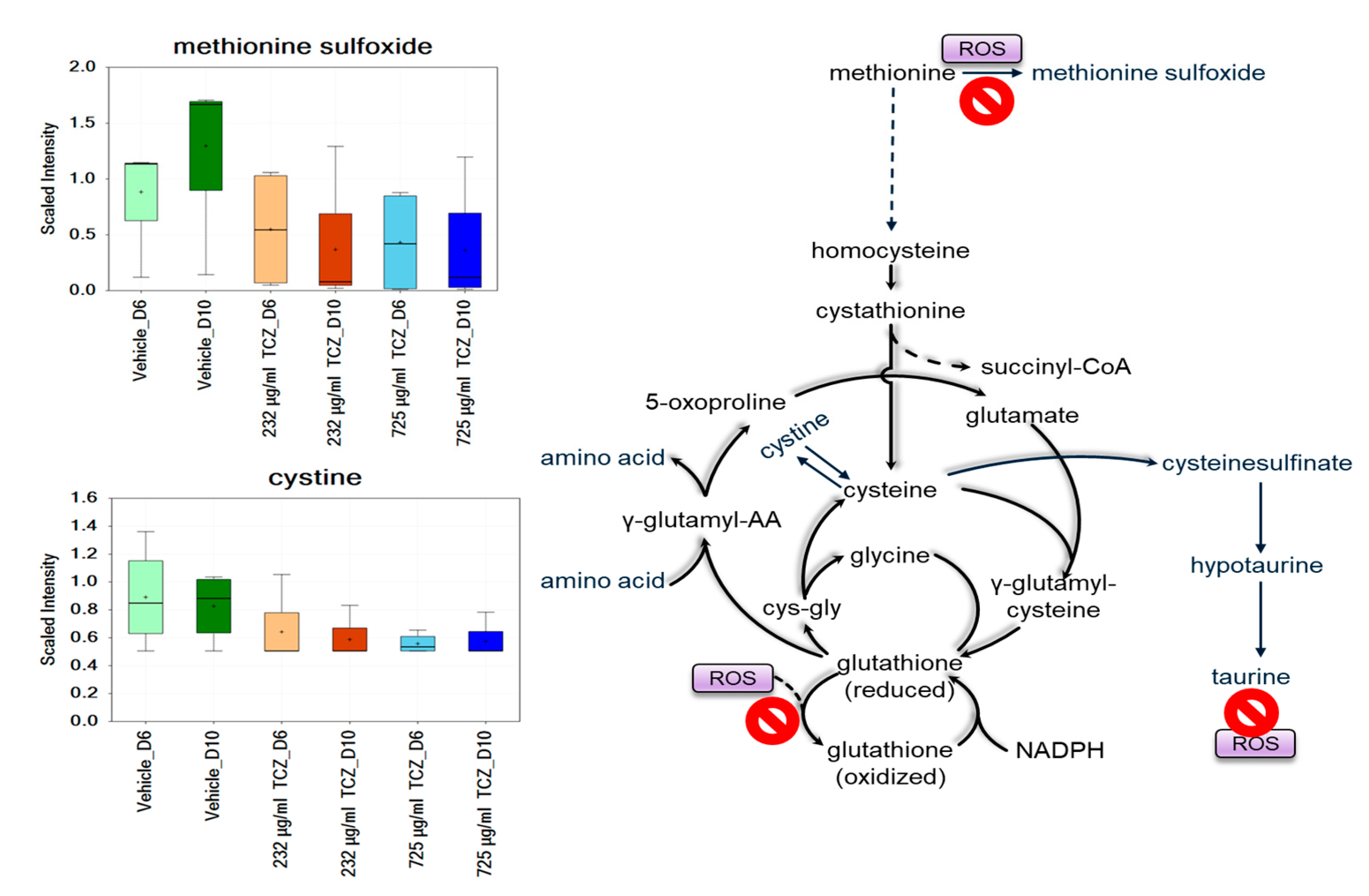
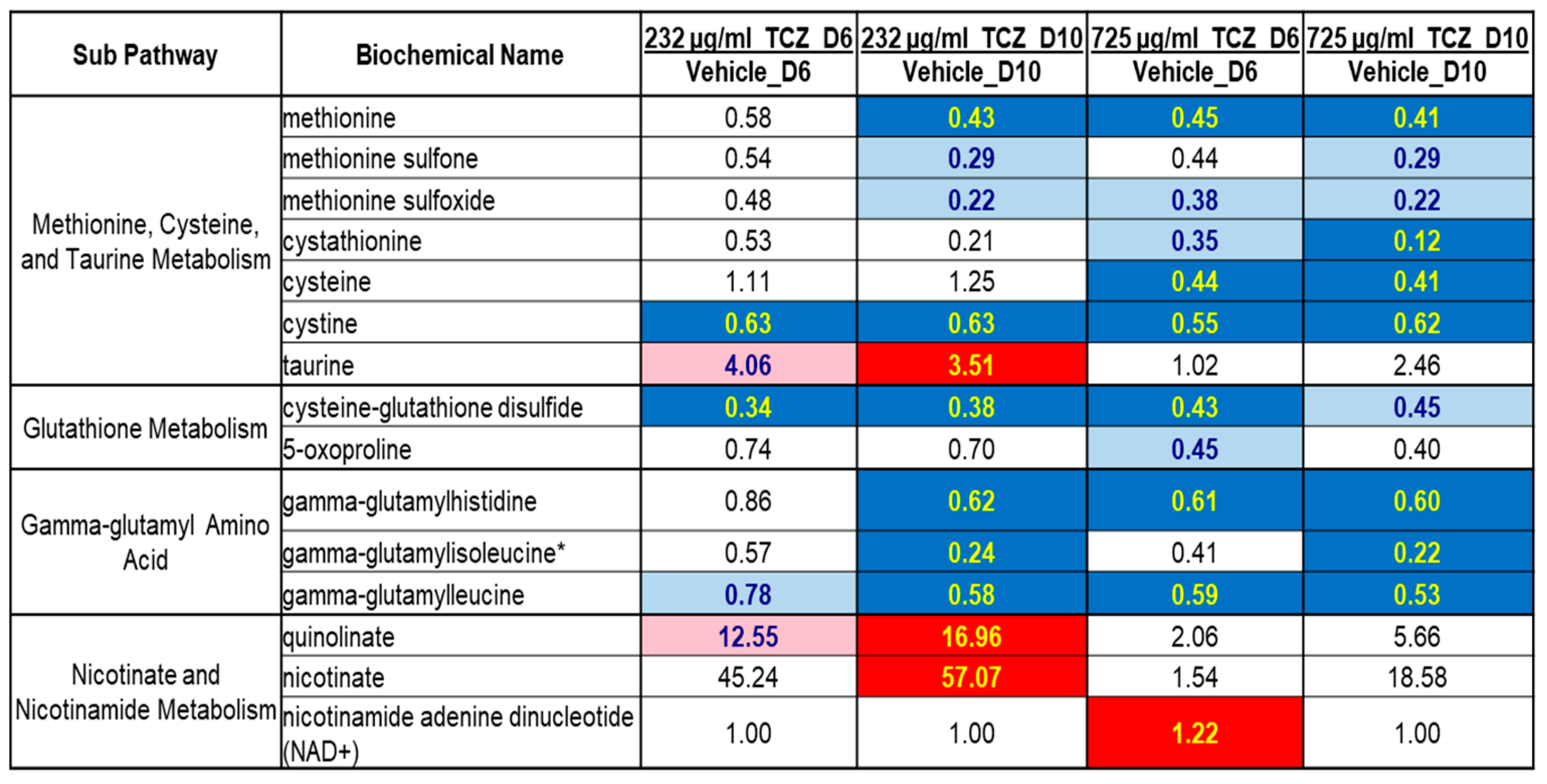
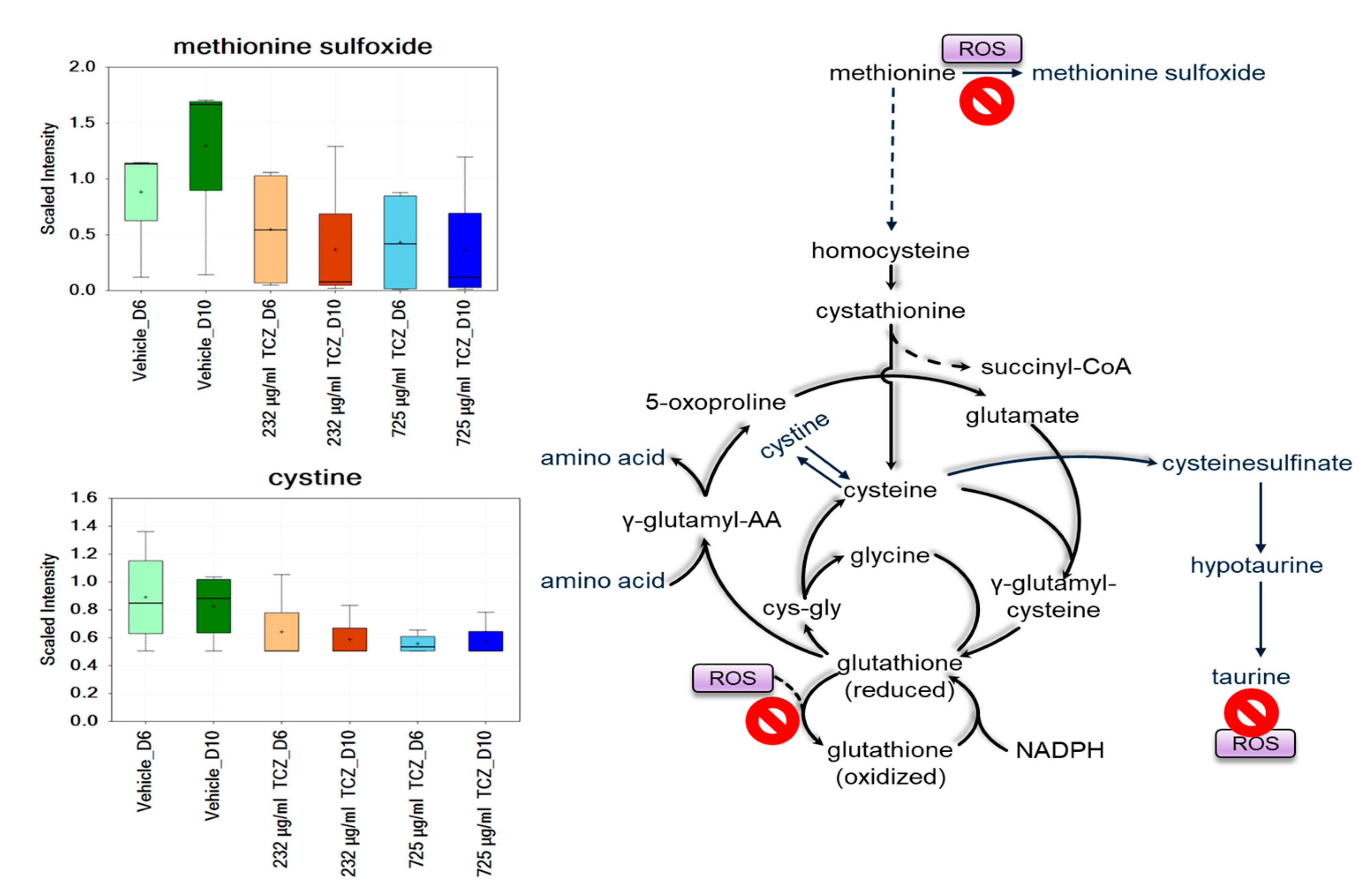
2.2. Metabolomics Analysis of Tocilizumab Effects in LAMPS Models
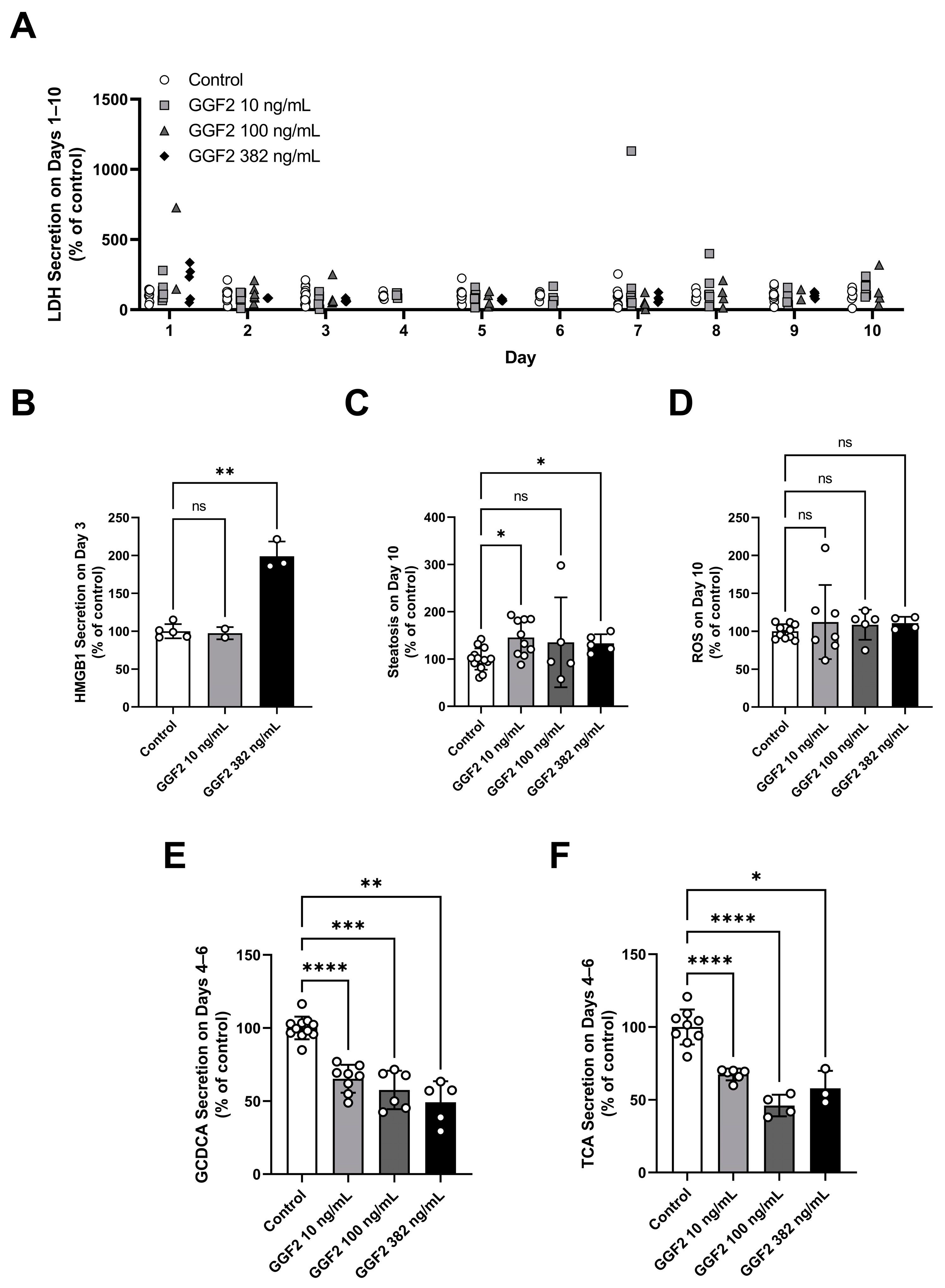
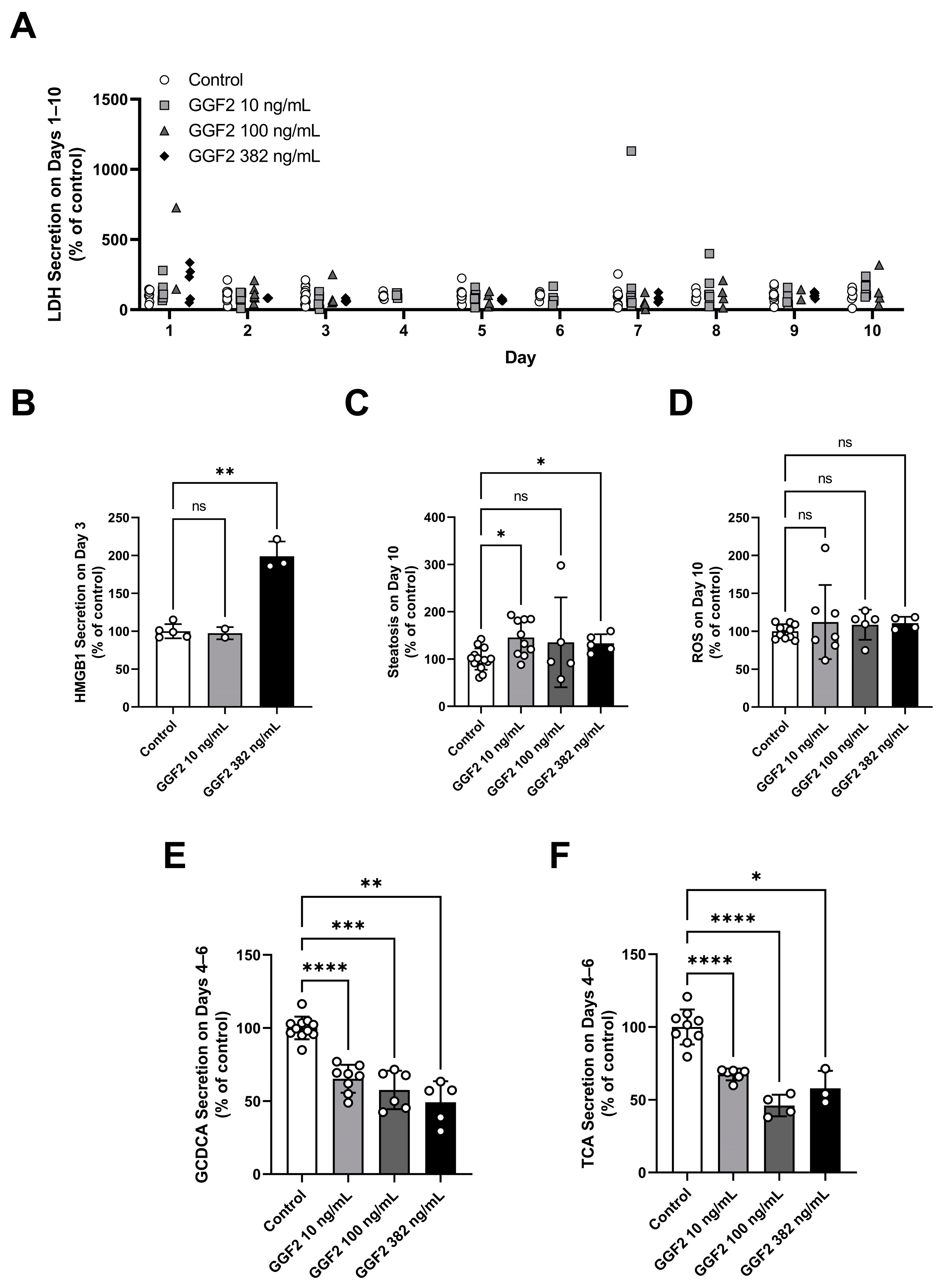
2.3. Evaluation of GGF2 Effects on Phenotypic and Mechanistic Toxicity Endpoints in LAMPS Models
2.4. Determination of Tocilizumab Mechanistic Toxicity Parameters
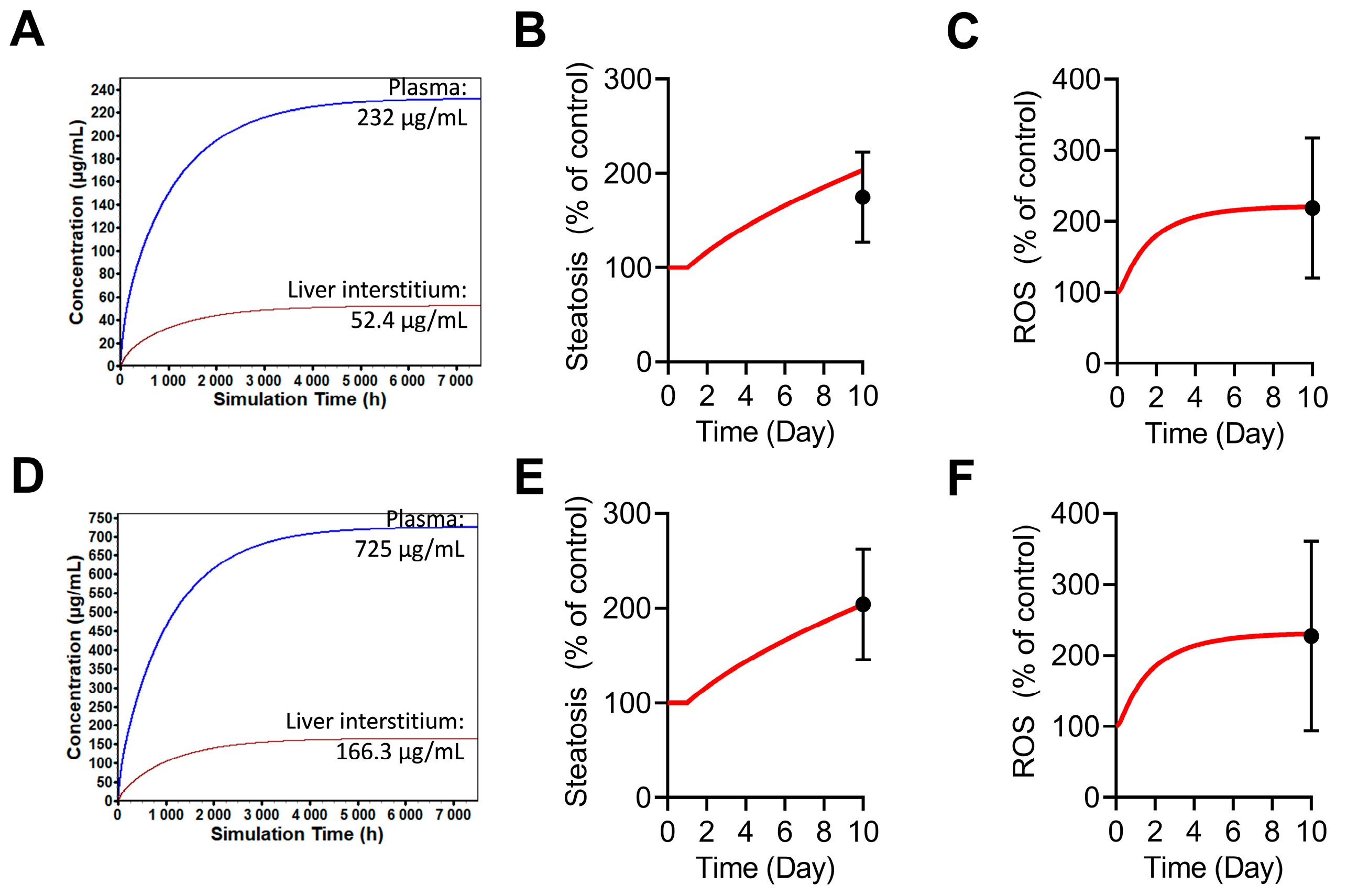
2.5. Simulations of Tocilizumab-Mediated Hepatotoxicity in BIOLOGXsym
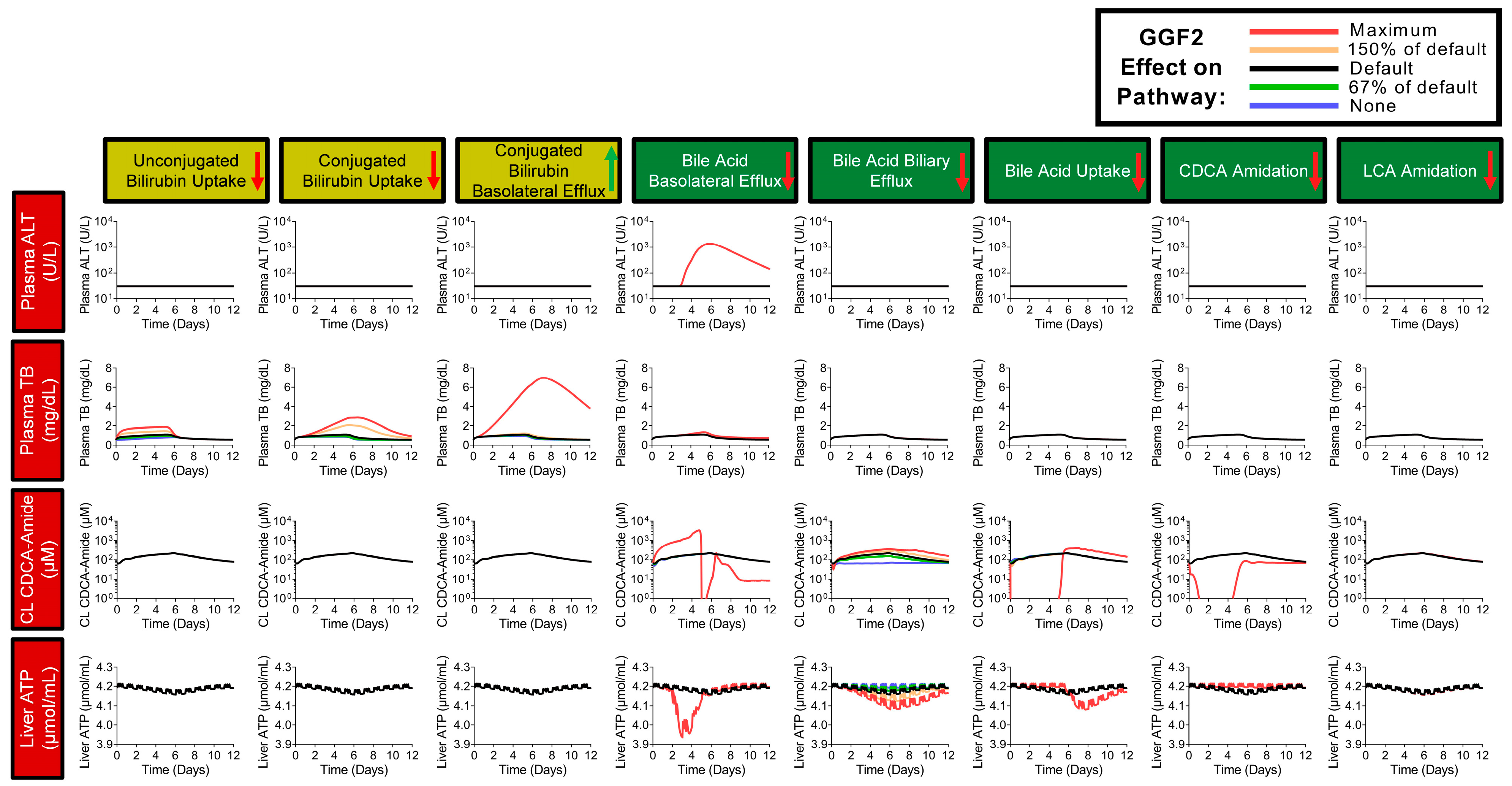
2.6. Determination of GGF2 Mechanistic Toxicity Parameters
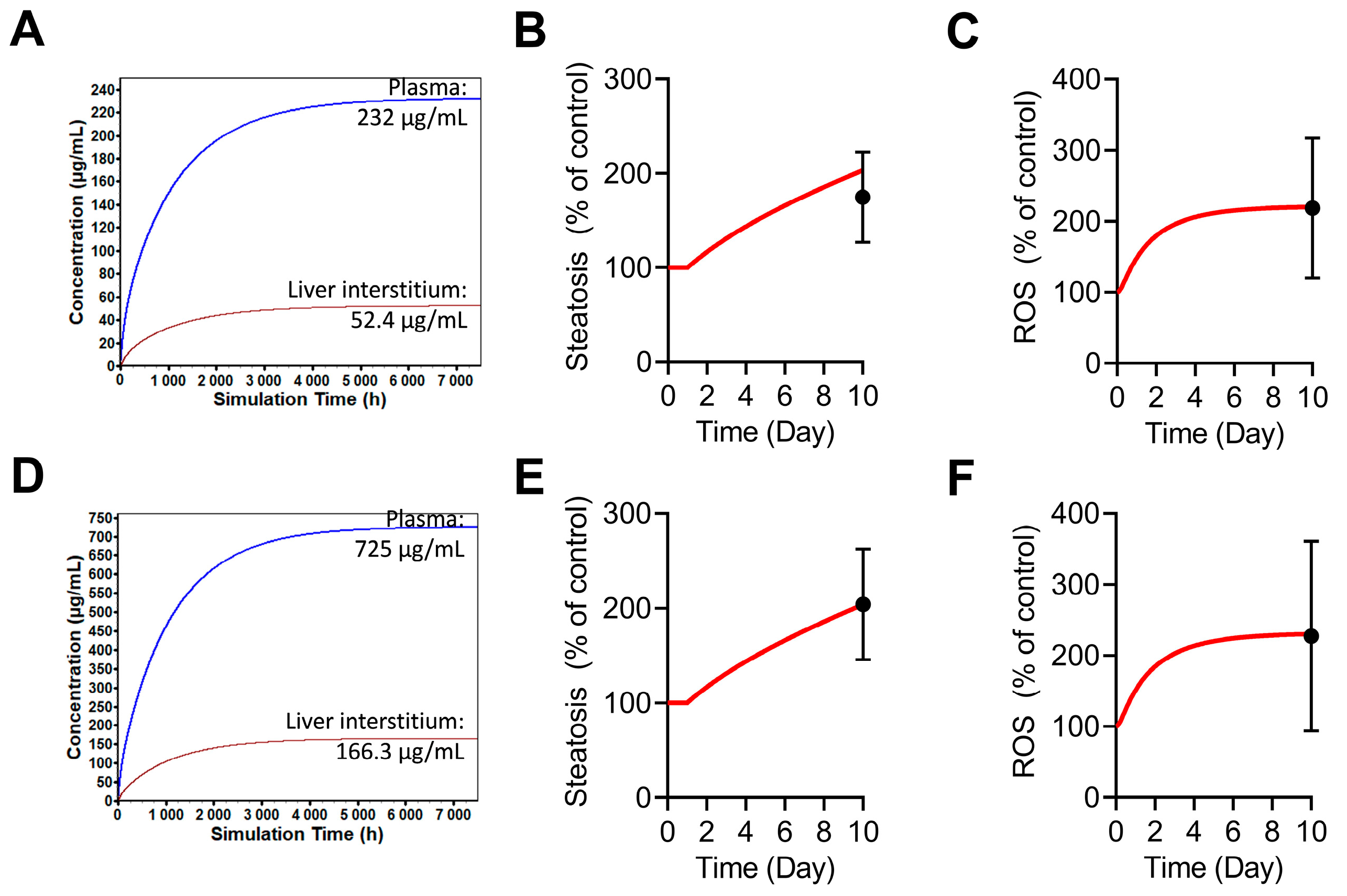
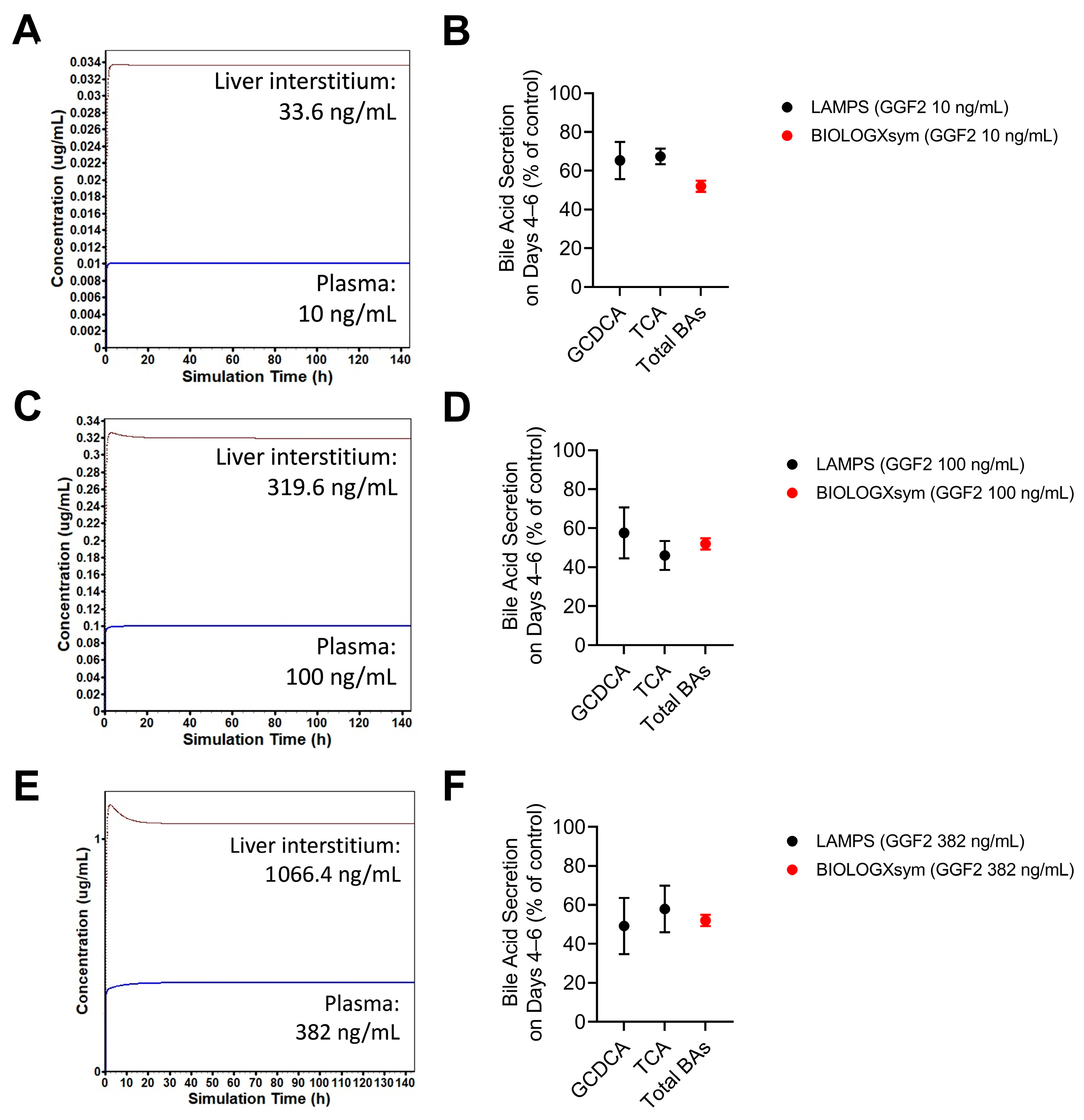
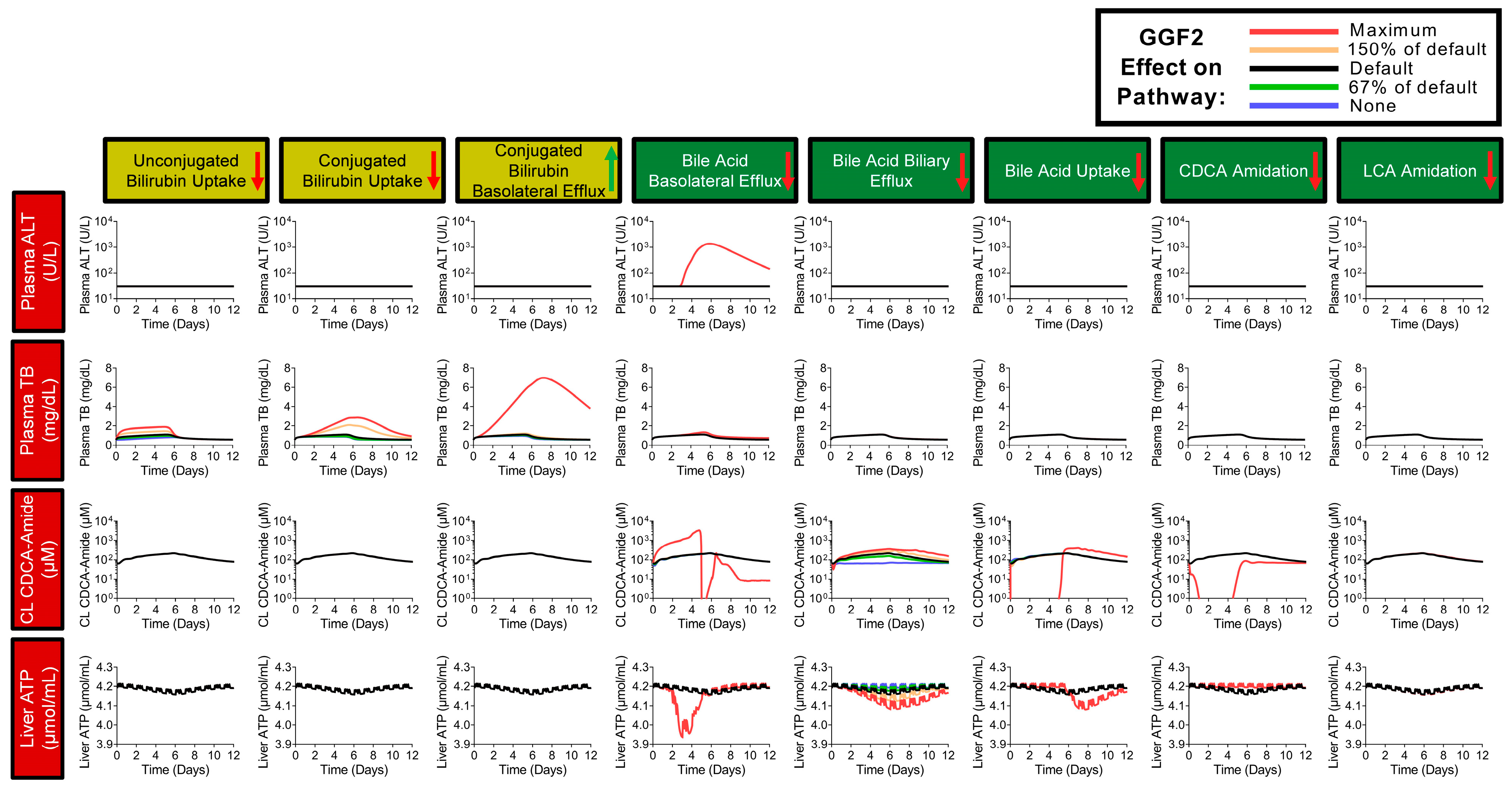
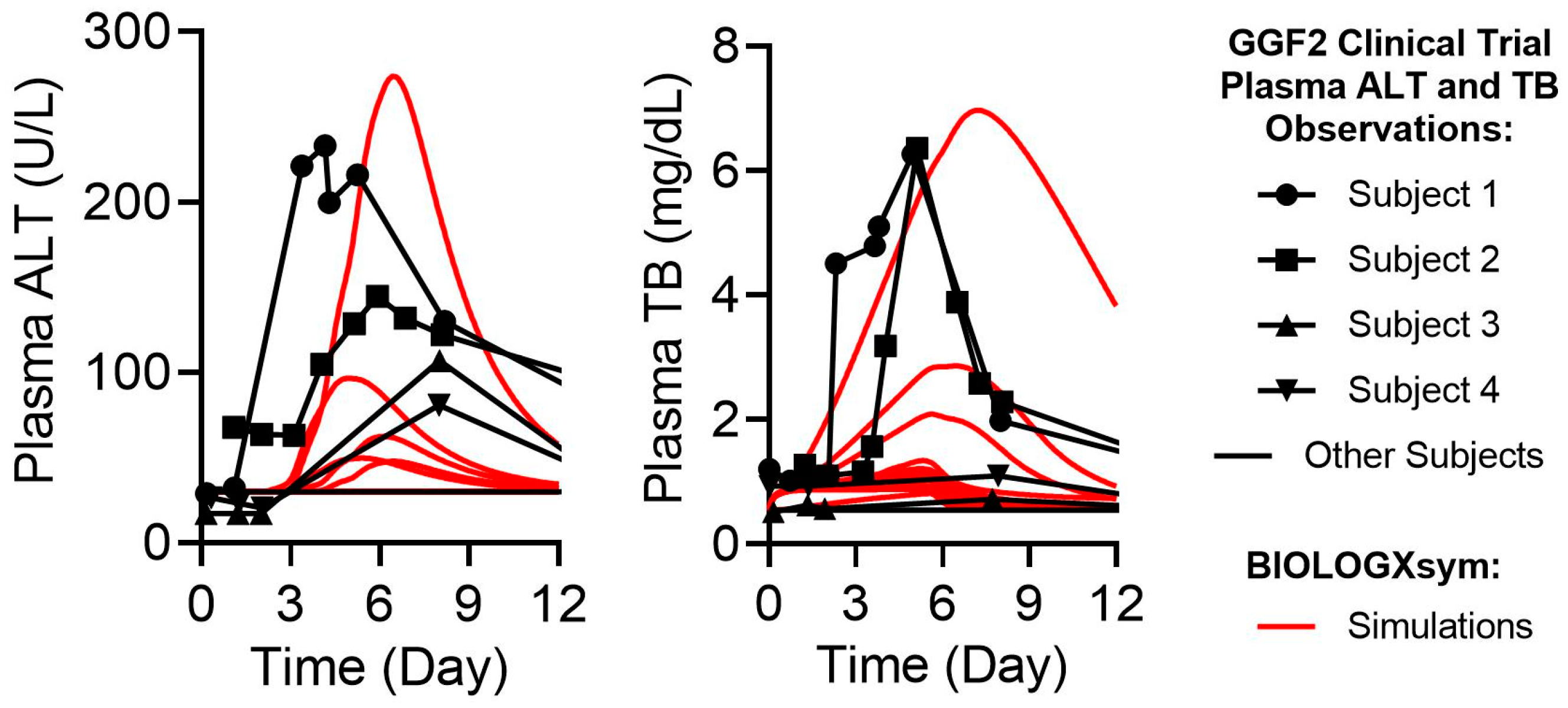
2.7. Simulations of GGF2-Mediated Hepatotoxicity in BIOLOGXsym
3. Discussion
4. Materials and Methods
4.1. The Liver Acinus Microphysiology System (LAMPS)
4.2. LAMPS Model Assembly and Maintenance
4.3. Normal-Fasting Media
4.4. Drug Preparation and Treatment
4.5. Device Fixation Protocol
4.6. Assessment of Drug Effects on Outcome Measurements in LAMPS
4.7. Statistical Analysis
4.8. BioSystics-Analytics Platform (BioSystics-AP)
4.9. Development of BIOLOGXsym
4.10. Development of Physiologically-Based Pharmacokinetic (PBPK) Models of Tocilizumab and GGF2
4.11. Hepatotoxicity Mechanisms and Parameters of Tocilizumab
4.12. Setup of Tocilizumab-Mediated Hepatotoxicity Simulations in BIOLOGXsym
4.13. Hepatotoxicity Mechanisms and Parameters of GGF2
4.14. Setup of GGF2-Mediated Hepatotoxicity Simulations in BIOLOGXsym
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- FDA. What Are “Biologics” Questions and Answers; US FDA: Silver Spring, MD, USA, 2018.
- Opportunities and Challenges in Biologic Drug Delivery. 2017. Available online: http://www.americanpharmaceuticalreview.com/Featured-Articles/345540-Opportunities-and-Challenges-in-Biologic-Drug-Delivery/ (accessed on 4 March 2020).
- Shah, P.; Sundaram, V.; Björnsson, E. Biologic and Checkpoint Inhibitor-Induced Liver Injury: A Systematic Literature Review. Hepatol. Commun. 2020, 4, 172–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosedale, M.; Button, D.; Jackson, J.P.; Freeman, K.M.; Brouwer, K.R.; Caggiano, A.O.; Eisen, A.; Iaci, J.F.; Parry, T.J.; Stanulis, R.; et al. Transient Changes in Hepatic Physiology That Alter Bilirubin and Bile Acid Transport May Explain Elevations in Liver Chemistries Observed in Clinical Trials of GGF2 (Cimaglermin Alfa). Toxicol. Sci. 2018, 161, 401–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, D.M.; Generaux, G.T.; Howell, B.A.; Siler, S.Q.; Antoine, D.J.; Button, D.; Caggiano, A.; Eisen, A.; Iaci, J.; Stanulis, R.; et al. Refining Liver Safety Risk Assessment: Application of Mechanistic Modeling and Serum Biomarkers to Cimaglermin Alfa (GGF2) Clinical Trials. Clin. Pharmacol. Ther. 2017, 102, 961–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenihan, D.J.; Anderson, S.A.; Lenneman, C.G.; Brittain, E.; Muldowney, J.A.S.; Mendes, L.; Zhao, P.Z.; Iaci, J.; Frohwein, S.; Zolty, R.; et al. A Phase I, Single Ascending Dose Study of Cimaglermin Alfa (Neuregulin 1β3) in Patients with Systolic Dysfunction and Heart Failure. JACC Basic. Transl. Sci. 2016, 1, 576–586. [Google Scholar] [CrossRef]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; van Vollenhoven, R.F. Integrated Safety in Tocilizumab Clinical Trials. Arthritis Res. Ther. 2011, 13, R141. [Google Scholar] [CrossRef] [Green Version]
- Anger, F.; Wiegering, A.; Wagner, J.; Lock, J.; Baur, J.; Haug, L.; Schmalzing, M.; Geier, A.; Löb, S.; Klein, I. Toxic Drug-Induced Liver Failure during Therapy of Rheumatoid Arthritis with Tocilizumab Subcutaneously: A Case Report. Rheumatology 2017, 56, 1628–1629. [Google Scholar] [CrossRef] [Green Version]
- Drepper, M.; Rubbia-Brandt, L.; Spahr, L. Tocilizumab-Induced Acute Liver Injury in Adult Onset Still’s Disease. Case Rep. Hepatol. 2013, 2013, 964828. [Google Scholar] [CrossRef]
- Mahamid, M.; Paz, K.; Reuven, M.; Safadi, R. Hepatotoxicity Due to Tocilizumab and Anakinra in Rheumatoid Arthritis: Two Case Reports. Int. J. Gen. Med. 2011, 4, 657–660. [Google Scholar] [CrossRef] [Green Version]
- Leach, M.W.; Clarke, O.D.; Dudal, S.; Han, C.; Li, C.; Yang, Z.; Brennan, F.R.; Bailey, W.J.; Chen, Y.; Deslandes, A.; et al. Strategies and Recommendations for Using a Data-Driven and Risk-Based Approach in the Selection of First-in-Human Starting Dose: An International Consortium for Innovation and Quality in Pharmaceutical Development (IQ) Assessment. Clin. Pharmacol. Ther. 2020, 109, 1395–1415. [Google Scholar] [CrossRef]
- Tibbitts, J.; Cavagnaro, J.A.; Haller, C.A.; Marafino, B.; Andrews, P.A.; Sullivan, J.T. Practical Approaches to Dose Selection for First-in-Human Clinical Trials with Novel Biopharmaceuticals. Regul. Toxicol. Pharmacol. 2010, 58, 243–251. [Google Scholar] [CrossRef]
- Vulto, P.; Joore, J. Adoption of Organ-on-Chip Platforms by the Pharmaceutical Industry. Nat. Rev. Drug. Discov. 2021, 20, 961–962. [Google Scholar] [CrossRef]
- Gough, A.; Soto-Gutierrez, A.; Vernetti, L.; Ebrahimkhani, M.R.; Stern, A.M.; Taylor, D.L. Human Biomimetic Liver Microphysiology Systems in Drug Development and Precision Medicine. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 252–268. [Google Scholar] [CrossRef]
- Vernetti, L.A.; Senutovitch, N.; Boltz, R.; DeBiasio, R.; Ying Shun, T.; Gough, A.; Taylor, D.L. A Human Liver Microphysiology Platform for Investigating Physiology, Drug Safety, and Disease Models. Exp. Biol. Med. 2016, 241, 101–114. [Google Scholar] [CrossRef]
- Lee-Montiel, F.T.; George, S.M.; Gough, A.H.; Sharma, A.D.; Wu, J.; DeBiasio, R.; Vernetti, L.A.; Taylor, D.L. Control of Oxygen Tension Recapitulates Zone-Specific Functions in Human Liver Microphysiology Systems. Exp. Biol. Med. 2017, 242, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Lefever, D.E.; Miedel, M.T.; Pei, F.; DiStefano, J.K.; Debiasio, R.; Shun, T.Y.; Saydmohammed, M.; Chikina, M.; Vernetti, L.A.; Soto-Gutierrez, A.; et al. A Quantitative Systems Pharmacology Platform Reveals NAFLD Pathophysiological States and Targeting Strategies. Metabolites 2022, 12, 528. [Google Scholar] [CrossRef]
- Stern, A.M.; Schurdak, M.E.; Bahar, I.; Berg, J.M.; Taylor, D.L. A Perspective on Implementing a Quantitative Systems Pharmacology Platform for Drug Discovery and the Advancement of Personalized Medicine. J. Biomol. Screen. 2016, 21, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.L.; Gough, A.; Schurdak, M.E.; Vernetti, L.; Chennubhotla, C.S.; Lefever, D.; Pei, F.; Faeder, J.R.; Lezon, T.R.; Stern, A.M.; et al. Harnessing Human Microphysiology Systems as Key Experimental Models for Quantitative Systems Pharmacology. Handb. Exp. Pharmacol. 2019, 260, 327–367. [Google Scholar] [CrossRef]
- Saydmohammed, M.; Jha, A.; Mahajan, V.; Gavlock, D.; Shun, T.Y.; DeBiasio, R.; Lefever, D.; Li, X.; Reese, C.; Kershaw, E.E.; et al. Quantifying the Progression of Non-Alcoholic Fatty Liver Disease in Human Biomimetic Liver Microphysiology Systems with Fluorescent Protein Biosensors. Exp. Biol. Med. 2021, 246, 2420–2441. [Google Scholar] [CrossRef]
- Bloomingdale, P.; Housand, C.; Apgar, J.F.; Millard, B.L.; Mager, D.E.; Burke, J.M.; Shah, D.K. Quantitative Systems Toxicology. Curr. Opin. Toxicol. 2017, 4, 79–87. [Google Scholar] [CrossRef]
- Watkins, P.B. Quantitative Systems Toxicology Approaches to Understand and Predict Drug-Induced Liver Injury. Clin. Liver Dis. 2020, 24, 49–60. [Google Scholar] [CrossRef]
- Smith, B.; Rowe, J.; Watkins, P.B.; Ashina, M.; Woodhead, J.L.; Sistare, F.D.; Goadsby, P.J. Mechanistic Investigations Support Liver Safety of Ubrogepant. Toxicol. Sci. 2020, 177, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Woodhead, J.L.; Yang, K.; Oldach, D.; MacLauchlin, C.; Fernandes, P.; Watkins, P.B.; Siler, S.Q.; Howell, B.A. Analyzing the Mechanisms Behind Macrolide Antibiotic-Induced Liver Injury Using Quantitative Systems Toxicology Modeling. Pharm. Res. 2019, 36, 48. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, J.J.; Brock, W.J.; Watkins, P.B.; Brouwer, K.L.R. Quantitative Systems Toxicology Modeling Predicts That Reduced Biliary Efflux Contributes to Tolvaptan Hepatotoxicity. Clin. Pharmacol. Ther. 2021, 109, 433–442. [Google Scholar] [CrossRef]
- Longo, D.M.; Woodhead, J.L.; Walker, P.; Herédi-Szabó, K.; Mogyorósi, K.; Wolenski, F.S.; Dragan, Y.P.; Mosedale, M.; Siler, S.Q.; Watkins, P.B.; et al. Quantitative Systems Toxicology Analysis of In Vitro Mechanistic Assays Reveals Importance of Bile Acid Accumulation and Mitochondrial Dysfunction in TAK-875-Induced Liver Injury. Toxicol. Sci. 2019, 167, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, T.J.; Cosgrove, P.A.; Dunn, R.T.; Stolz, D.B.; Hamadeh, H.; Afshari, C.; McBride, H.; Griffith, L.G. Modeling Therapeutic Antibody-Small Molecule Drug-Drug Interactions Using a Three-Dimensional Perfusable Human Liver Coculture Platform. Drug. Metab. Dispos. 2016, 44, 1940–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurdak, M.; Vernetti, L.; Bergenthal, L.; Wolter, Q.K.; Shun, T.Y.; Karcher, S.; Taylor, D.L.; Gough, A. Applications of the Microphysiology Systems Database for Experimental ADME-Tox and Disease Models. Lab. Chip. 2020, 20, 1472–1492. [Google Scholar] [CrossRef] [Green Version]
- Church, R.J.; Watkins, P.B. The Transformation in Biomarker Detection and Management of Drug-Induced Liver Injury. Liver Int. 2017, 37, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, F.; Liang, B.; Su, Y.; Sun, S.; Xia, S.; Shao, J.; Zhang, Z.; Hong, M.; Zhang, F.; et al. Methionine Metabolism in Chronic Liver Diseases: An Update on Molecular Mechanism and Therapeutic Implication. Signal. Transduct. Target. Ther. 2020, 5, 280. [Google Scholar] [CrossRef]
- Lee, B.C.; Gladyshev, V.N. The Biological Significance of Methionine Sulfoxide Stereochemistry. Free Radic. Biol. Med. 2011, 50, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal. Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, J.; Yin, Y.; Li, K.; Zhang, C.; Zheng, Y. The Role of IL-6 in Fibrotic Diseases: Molecular and Cellular Mechanisms. Int. J. Biol. Sci. 2022, 18, 5405–5414. [Google Scholar] [CrossRef]
- Gough, A.; Vernetti, L.; Bergenthal, L.; Shun, T.Y.; Taylor, D.L. The Microphysiology Systems Database for Analyzing and Modeling Compound Interactions with Human and Animal Organ Models. Appl. Vitr. Toxicol. 2016, 2, 103–117. [Google Scholar] [CrossRef]
- Blomme, E.A.G.; Will, Y. Toxicology Strategies for Drug Discovery: Present and Future. Chem. Res. Toxicol. 2016, 29, 473–504. [Google Scholar] [CrossRef]
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human Drug-Induced Liver Injury Severity Is Highly Associated with Dual Inhibition of Liver Mitochondrial Function and Bile Salt Export Pump. Hepatology 2014, 60, 1015–1022. [Google Scholar] [CrossRef]
- Pereira, C.V.; Nadanaciva, S.; Oliveira, P.J.; Will, Y. The Contribution of Oxidative Stress to Drug-Induced Organ Toxicity and Its Detection in Vitro and in Vivo. Expert. Opin. Drug. Metab. Toxicol. 2012, 8, 219–237. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Jakubczyk, K.; Dec, K.; Kałduńska, J.; Kawczuga, D.; Kochman, J.; Janda, K. Reactive Oxygen Species–Sources, Functions, Oxidative Damage. Pol. Merkur. Lekarski 2020, 48, 124–127. [Google Scholar]
- D’Alessandro, A.; Rinalducci, S.; Zolla, L. Redox Proteomics and Drug Development. J. Proteom. 2011, 74, 2575–2595. [Google Scholar] [CrossRef]
- Hoshi, T.; Heinemann, S. Regulation of Cell Function by Methionine Oxidation and Reduction. J. Physiol. 2001, 531, 1–11. [Google Scholar] [CrossRef]
- Banerjee, R. Redox Outside the Box: Linking Extracellular Redox Remodeling with Intracellular Redox Metabolism. J. Biol. Chem. 2012, 287, 4397–4402. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, D.A.; Forman, H.J. Cellular Glutathione and Thiols Metabolism. Biochem. Pharmacol. 2002, 64, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of Hepatic Glutathione Synthesis: Current Concepts and Controversies. FASEB J. 1999, 13, 1169–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappaport, B. Tocilizumab Summary Review; FDA Center for Drug Evaluation and Research: Beltsville, MD, USA, 2009.
- Fazel Modares, N.; Polz, R.; Haghighi, F.; Lamertz, L.; Behnke, K.; Zhuang, Y.; Kordes, C.; Häussinger, D.; Sorg, U.R.; Pfeffer, K.; et al. IL-6 Trans-Signaling Controls Liver Regeneration After Partial Hepatectomy. Hepatology 2019, 70, 2075–2091. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Rose-John, S. IL-6 Pathway in the Liver: From Physiopathology to Therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.; Blinn, G.; Jostock, T.; Schirmacher, P.; Meyer zum Büschenfelde, K.H.; Galle, P.R.; Rose-John, S. Combined Interleukin 6 and Soluble Interleukin 6 Receptor Accelerates Murine Liver Regeneration. Gastroenterology 2000, 119, 1663–1671. [Google Scholar] [CrossRef]
- Lin, Z.-Q.; Kondo, T.; Ishida, Y.; Takayasu, T.; Mukaida, N. Essential Involvement of IL-6 in the Skin Wound-Healing Process as Evidenced by Delayed Wound Healing in IL-6-Deficient Mice. J. Leukoc. Biol. 2003, 73, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Lindbom, L.; Weber, C. Mechanisms Underlying Neutrophil-Mediated Monocyte Recruitment. Blood 2009, 114, 4613–4623. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.; Scuderi, F.; Provenzano, C.; Scheller, J.; Rose-John, S.; Bartoccioni, E. IL-6 Regulates MCP-1, ICAM-1 and IL-6 Expression in Human Myoblasts. J. Neuroimmunol. 2008, 196, 41–48. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The Pro- and Anti-Inflammatory Properties of the Cytokine Interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Jover, R.; Bort, R.; Gómez-Lechón, M.J.; Castell, J.V. Down-Regulation of Human CYP3A4 by the Inflammatory Signal Interleukin-6: Molecular Mechanism and Transcription Factors Involved. FASEB J. 2002, 16, 1799–1801. [Google Scholar] [CrossRef]
- Dickmann, L.J.; Patel, S.K.; Rock, D.A.; Wienkers, L.C.; Slatter, J.G. Effects of Interleukin-6 (IL-6) and an Anti-IL-6 Monoclonal Antibody on Drug-Metabolizing Enzymes in Human Hepatocyte Culture. Drug. Metab. Dispos. 2011, 39, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hao, C.; Yang, D.; Shi, D.; Song, X.; Luan, X.; Hu, G.; Yan, B. Pregnane X Receptor Is Required for Interleukin-6-Mediated down-Regulation of Cytochrome P450 3A4 in Human Hepatocytes. Toxicol. Lett. 2010, 197, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Siewert, E.; Bort, R.; Kluge, R.; Heinrich, P.C.; Castell, J.; Jover, R. Hepatic Cytochrome P450 Down-Regulation during Aseptic Inflammation in the Mouse Is Interleukin 6 Dependent. Hepatology 2000, 32, 49–55. [Google Scholar] [CrossRef]
- Li, S.-Q.; Zhu, S.; Han, H.-M.; Lu, H.-J.; Meng, H.-Y. IL-6 Trans-Signaling Plays Important Protective Roles in Acute Liver Injury Induced by Acetaminophen in Mice. J. Biochem. Mol. Toxicol. 2015, 29, 288–297. [Google Scholar] [CrossRef]
- Hakkola, J.; Hu, Y.; Ingelman-Sundberg, M. Mechanisms of Down-Regulation of CYP2E1 Expression by Inflammatory Cytokines in Rat Hepatoma Cells. J. Pharmacol. Exp. Ther. 2003, 304, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, N.; Kishimoto, T. Humanized Antihuman IL-6 Receptor Antibody, Tocilizumab. Handb. Exp. Pharmacol. 2008, 181, 151–160. [Google Scholar] [CrossRef]
- Mihara, M.; Kasutani, K.; Okazaki, M.; Nakamura, A.; Kawai, S.; Sugimoto, M.; Matsumoto, Y.; Ohsugi, Y. Tocilizumab Inhibits Signal Transduction Mediated by Both MIL-6R and SIL-6R, but Not by the Receptors of Other Members of IL-6 Cytokine Family. Int. Immunopharmacol. 2005, 5, 1731–1740. [Google Scholar] [CrossRef]
- Chiew, A.L.; Buckley, N.A. Acetaminophen Poisoning. Crit. Care Clin. 2021, 37, 543–561. [Google Scholar] [CrossRef]
- Ghany, M.G.; Watkins, P.B. Moving the Needle to Reduce Acetaminophen (Paracetamol) Hepatotoxicity. JAMA 2023, 329, 713–715. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights Into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef]
- Murphy, W.A.; Adiwidjaja, J.; Sjöstedt, N.; Yang, K.; Beaudoin, J.J.; Spires, J.; Siler, S.Q.; Neuhoff, S.; Brouwer, K.L.R. Considerations for Physiologically Based Modeling in Liver Disease: From Nonalcoholic Fatty Liver (NAFL) to Nonalcoholic Steatohepatitis (NASH). Clin. Pharmacol. Ther. 2023, 113, 275–297. [Google Scholar] [CrossRef] [PubMed]
- El-Khateeb, E.; Achour, B.; Scotcher, D.; Al-Majdoub, Z.M.; Athwal, V.; Barber, J.; Rostami-Hodjegan, A. Scaling Factors for Clearance in Adult Liver Cirrhosis. Drug. Metab. Dispos. 2020, 48, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Docci, L.; Milani, N.; Ramp, T.; Romeo, A.A.; Godoy, P.; Franyuti, D.O.; Krähenbühl, S.; Gertz, M.; Galetin, A.; Parrott, N.; et al. Exploration and Application of a Liver-on-a-Chip Device in Combination with Modelling and Simulation for Quantitative Drug Metabolism Studies. Lab. Chip 2022, 22, 1187–1205. [Google Scholar] [CrossRef] [PubMed]
- Vernetti, L.; Gough, A.; Baetz, N.; Blutt, S.; Broughman, J.R.; Brown, J.A.; Foulke-Abel, J.; Hasan, N.; In, J.; Kelly, E.; et al. Corrigendum: Functional Coupling of Human Microphysiology Systems: Intestine, Liver, Kidney Proximal Tubule, Blood-Brain Barrier and Skeletal Muscle. Sci. Rep. 2017, 7, 44517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colnot, S.; Perret, C. Liver Zonation. In Molecular Pathology of Liver Diseases; Monga, S.P.S., Ed.; Molecular Pathology Library; Springer: Boston, MA, USA, 2011; pp. 7–16. ISBN 978-1-4419-7107-4. [Google Scholar]
- Wikswo, J.P.; Curtis, E.L.; Eagleton, Z.E.; Evans, B.C.; Kole, A.; Hofmeister, L.H.; Matloff, W.J. Scaling and Systems Biology for Integrating Multiple Organs-on-a-Chip. Lab. Chip 2013, 13, 3496–3511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Kaplowitz, N. Mechanisms of Drug-Induced Liver Injury. Clin. Liver Dis. 2013, 17, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Woodhead, J.L.; Howell, B.A.; Yang, Y.; Harrill, A.H.; Clewell, H.J., 3rd; Andersen, M.E.; Siler, S.Q.; Watkins, P.B. An Analysis of N-Acetylcysteine Treatment for Acetaminophen Overdose Using a Systems Model of Drug-Induced Liver Injury. J. Pharmacol. Exp. Ther. 2012, 342, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Howell, B.A.; Yang, Y.; Kumar, R.; Woodhead, J.L.; Harrill, A.H.; Clewell, H.J., 3rd; Andersen, M.E.; Siler, S.Q.; Watkins, P.B. In Vitro to in Vivo Extrapolation and Species Response Comparisons for Drug-Induced Liver Injury (DILI) Using DILIsymTM: A Mechanistic, Mathematical Model of DILI. J. Pharmacokinet. Pharmacodyn. 2012, 39, 527–541. [Google Scholar] [CrossRef]
- Howell, B.A.; Siler, S.Q.; Watkins, P.B. Use of a Systems Model of Drug-Induced Liver Injury (DILIsym(®)) to Elucidate the Mechanistic Differences between Acetaminophen and Its Less-Toxic Isomer, AMAP, in Mice. Toxicol. Lett. 2014, 226, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.; Bridgewater, B.; Liu, Q.; Mitchel, A.; Robinson, R. High Resolution Mass Spectrometry Improves Data Quantity and Quality as Compared to Unit Mass Resolution Mass Spectrometry in High-Throughput Profiling Metabolomics. J. Postgenom. Drug Biomark. Dev. 2014, 4, 132. [Google Scholar] [CrossRef] [Green Version]
- DeHaven, C.D.; Evans, A.M.; Dai, H.; Lawton, A.K. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Chemin 2010, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Woodhead, J.L.; Yang, K.; Siler, S.Q.; Watkins, P.B.; Brouwer, K.L.R.; Barton, H.A.; Howell, B.A. Exploring BSEP inhibition-mediated toxicity with a mechanistic model of drug-induced liver injury. Front. Pharmacol. 2014, 5, 240. [Google Scholar] [CrossRef] [Green Version]
- Woodhead, J.; Yang, K.; Brouwer, K.; Siler, S.; Stahl, S.; Ambroso, J.; Baker, D.; Watkins, P.; Howell, B. Mechanistic Modeling Reveals the Critical Knowledge Gaps in Bile Acid-Mediated DILI. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 123–128. [Google Scholar] [CrossRef]
- Balmagiya, T.; Rozovski, S.J. Thermoregulation in Young Adult Rats during Short-and Long-Term Protein Malnutrition. J. Nutr. 1983, 113, 228–238. [Google Scholar] [CrossRef]
- Ussher, J.R.; Koves, T.R.; Cadete, V.J.; Zhang, L.; Jaswal, J.S.; Swyrd, S.J.; Lopaschuk, D.G.; Proctor, S.D.; Keung, W.; Muoio, D.M.; et al. Inhibition of De Novo Ceramide Synthesis Reverses Diet-Induced Insulin Resistance and Enhances Whole-Body Oxygen Consumption. Diabetes 2010, 59, 2453–2464. [Google Scholar] [CrossRef] [Green Version]
- Gonzales-Pacheco, D.M.; Buss, W.C.; Koehler, K.M.; Woodside, W.F.; Alpert, S.S. Energy Restriction Reduces Metabolic Rate in Adult Male Fisher-344 Rats. J. Nutr. 1993, 123, 90–97. [Google Scholar] [CrossRef]
- Lazzer, S.; Bedogni, G.; Lafortuna, C.; Marazzi, N.; Busti, C.; Galli, R.; De Col, A.; Agosti, F.; Sartorio, A. Relationship Between Basal Metabolic Rate, Gender, Age, and Body Composition in 8,780 White Obese Subjects. Obesity 2010, 18, 71–78. [Google Scholar] [CrossRef]
- Wang, Z.; Ying, Z.; Bosy-Westphal, A.; Zhang, J.; Heller, M.; Later, W.; Heymsfield, S.B.; Müller, M.J. Evaluation of specific metabolic rates of major organs and tissues: Comparison between men and women. Am. J. Hum. Biol. 2010, 23, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.R. Computer-based Method for Calculation of the Available Energy of Proteins. J. Nutr. 1975, 105, 200–207. [Google Scholar] [CrossRef]
- Howell, B.; Siler, S.; Shoda, L.; Yang, Y.; Woodhead, J.; Watkins, P. A Mechanistic Model of Drug-Induced Liver Injury Aids the Interpretation of Elevated Liver Transaminase Levels in a Phase I Clinical Trial. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 1–8. [Google Scholar] [CrossRef]
- Yang, K.; Battista, C.; Woodhead, J.; Stahl, S.; Mettetal, J.; Watkins, P.; Siler, S.; Howell, B. Systems pharmacology modeling of drug-induced hyperbilirubinemia: Differentiating hepatotoxicity and inhibition of enzymes/transporters. Clin. Pharmacol. Ther. 2017, 101, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6·soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 2018, 293, 6762–6775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoda, L.K.; Battista, C.; Siler, S.Q.; Pisetsky, D.S.; Watkins, P.B.; Howell, A.B. Mechanistic Modelling of Drug-Induced Liver Injury: Investigating the Role of Innate Immune Responses. Gene Regul. Syst. Biol. 2017, 11, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef]
- Glassman, P.M.; Chen, Y.; Balthasar, J.P. Scale-up of a physiologically-based pharmacokinetic model to predict the disposition of monoclonal antibodies in monkeys. J. Pharmacokinet. Pharmacodyn. 2015, 42, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Georgy, A.; Rowell, L. Pharmacokinetics and pharmacodynamics of tocilizumab, a humanized anti-interleukin-6 receptor monoclonal antibody, following single-dose administration by subcutaneous and intravenous routes to healthy subjects. Int. J. Clin. Pharmacol. Ther. 2013, 51, 443–455. [Google Scholar] [CrossRef]
- Abdallah, H.; Hsu, J.C.; Lu, P.; Fettner, S.; Zhang, X.; Douglass, W.; Bao, M.; Rowell, L.; Burmester, G.R.; Kivitz, A. Pharmacokinetic and Pharmacodynamic Analysis of Subcutaneous Tocilizumab in Patients with Rheumatoid Arthritis From 2 Randomized, Controlled Trials: SUMMACTA and BREVACTA. J. Clin. Pharmacol. 2016, 57, 459–468. [Google Scholar] [CrossRef]
- Abou-Auda, H.S.; Sakr, W. Tocilizumab: A new anti-rheumatic drug. Saudi Pharm. J. 2010, 18, 257–259. [Google Scholar] [CrossRef] [Green Version]
- Parry, T.J.; Ganguly, A.; Troy, E.L.; Guerrero, J.L.; Iaci, J.F.; Srinivas, M.; Vecchione, A.M.; Button, D.C.; Hackett, C.S.; Zolty, R.; et al. Effects of neuregulin GGF2 (cimaglermin alfa) dose and treatment frequency on left ventricular function in rats following myocardial infarction. Eur. J. Pharmacol. 2017, 796, 76–89. [Google Scholar] [CrossRef]
- Baulida, J.; Kraus, M.H.; Alimandi, M.; Di Fiore, P.P.; Carpenter, G. All ErbB Receptors Other Than the Epidermal Growth Factor Receptor Are Endocytosis Impaired. J. Biol. Chem. 1996, 271, 5251–5257. [Google Scholar] [CrossRef] [Green Version]
- Long, C.L.; Dillard, D.R.; Bodzin, J.H.; Geiger, J.W.; Blakemore, W.S. Validity of 3-methylhistidine excretion as an indicator of skeletal muscle protein breakdown in humans. Metabolism 1988, 37, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.N.G.D.S.; Paula, D.P.; de Araujo, B.P.; Fonseca, M.D.J.M.D.; Diniz, M.D.F.H.S.; Daliry, A.; Griep, R.H. Advanced glycation end product: A potential biomarker for risk stratification of non-alcoholic fatty liver disease in ELSA-Brasil study. World J. Gastroenterol. 2021, 27, 4913–4928. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaudoin, J.J.; Clemens, L.; Miedel, M.T.; Gough, A.; Zaidi, F.; Ramamoorthy, P.; Wong, K.E.; Sarangarajan, R.; Battista, C.; Shoda, L.K.M.; et al. The Combination of a Human Biomimetic Liver Microphysiology System with BIOLOGXsym, a Quantitative Systems Toxicology (QST) Modeling Platform for Macromolecules, Provides Mechanistic Understanding of Tocilizumab- and GGF2-Induced Liver Injury. Int. J. Mol. Sci. 2023, 24, 9692. https://doi.org/10.3390/ijms24119692
Beaudoin JJ, Clemens L, Miedel MT, Gough A, Zaidi F, Ramamoorthy P, Wong KE, Sarangarajan R, Battista C, Shoda LKM, et al. The Combination of a Human Biomimetic Liver Microphysiology System with BIOLOGXsym, a Quantitative Systems Toxicology (QST) Modeling Platform for Macromolecules, Provides Mechanistic Understanding of Tocilizumab- and GGF2-Induced Liver Injury. International Journal of Molecular Sciences. 2023; 24(11):9692. https://doi.org/10.3390/ijms24119692
Chicago/Turabian StyleBeaudoin, James J., Lara Clemens, Mark T. Miedel, Albert Gough, Fatima Zaidi, Priya Ramamoorthy, Kari E. Wong, Rangaprasad Sarangarajan, Christina Battista, Lisl K. M. Shoda, and et al. 2023. "The Combination of a Human Biomimetic Liver Microphysiology System with BIOLOGXsym, a Quantitative Systems Toxicology (QST) Modeling Platform for Macromolecules, Provides Mechanistic Understanding of Tocilizumab- and GGF2-Induced Liver Injury" International Journal of Molecular Sciences 24, no. 11: 9692. https://doi.org/10.3390/ijms24119692
APA StyleBeaudoin, J. J., Clemens, L., Miedel, M. T., Gough, A., Zaidi, F., Ramamoorthy, P., Wong, K. E., Sarangarajan, R., Battista, C., Shoda, L. K. M., Siler, S. Q., Taylor, D. L., Howell, B. A., Vernetti, L. A., & Yang, K. (2023). The Combination of a Human Biomimetic Liver Microphysiology System with BIOLOGXsym, a Quantitative Systems Toxicology (QST) Modeling Platform for Macromolecules, Provides Mechanistic Understanding of Tocilizumab- and GGF2-Induced Liver Injury. International Journal of Molecular Sciences, 24(11), 9692. https://doi.org/10.3390/ijms24119692