
Methotrexate Provokes Disparate Folate Metabolism Gene Expression and Alternative Splicing in Ex Vivo Monocytes and GM-CSF- and M-CSF-Polarized Macrophages

 , ,
, ,  , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. FPGS Pre-mRNA Splicing, FPGS Activity and MTX-Polyglutamylation in Monocytes and M1- and M2-Type Macrophages
2.2. Differential Splicing Profiles in Monocytes, M1–MØ and M2–MØ
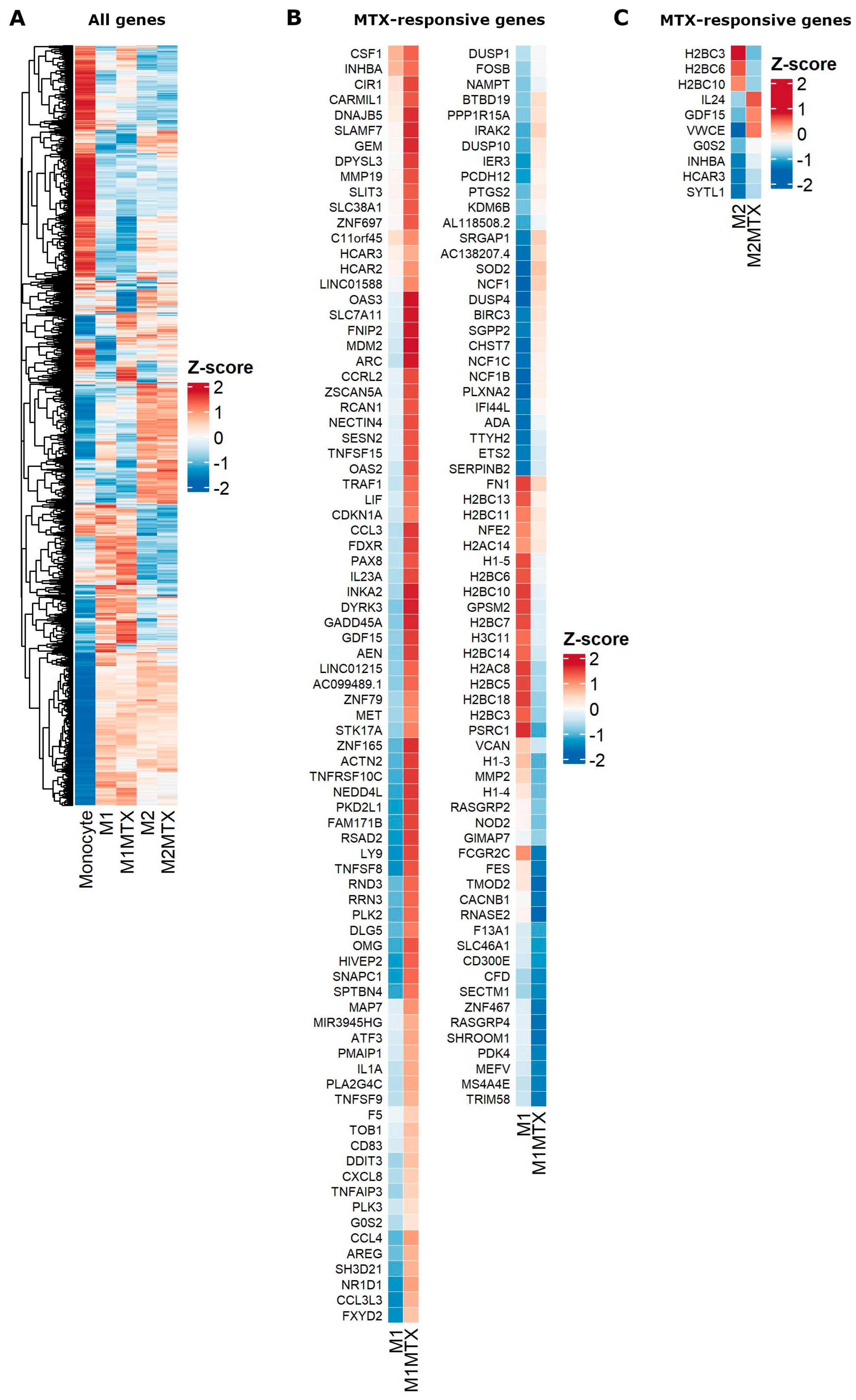
2.3. Differential Gene Expression (DGE) Profiling of Monocytes, M1–MØ and M2–MØ (with and without MTX Exposure)
3. Discussion
4. Materials and Methods
4.1. Materials
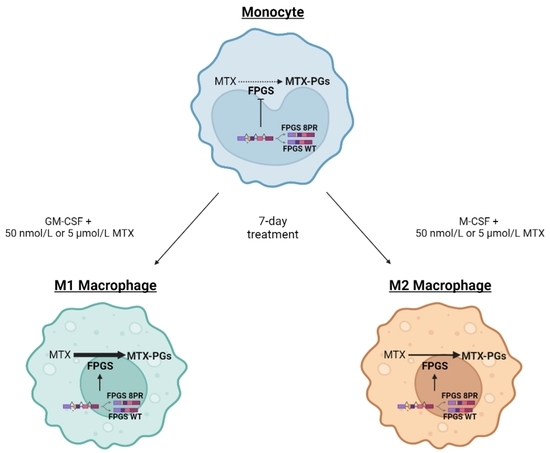
4.2. Monocyte-Macrophage Polarization Studies
4.3. RNA Isolation and RNA-Seq
4.4. RNA-Seq Analysis
4.5. qPCR Analysis
4.6. FPGS Catalytic Assay
4.7. LC-MS/MS MTX-PG Analysis
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.L.; Van Haandel, L.; Gaedigk, R.; Lasky, A.; Hoeltzel, M.; Stobaugh, J.; Leeder, J.S. Analysis of intracellular methotrexate polyglutamates in patients with juvenile idiopathic arthritis: Effect of route of administration on variability in intracellular methotrexate polyglutamate concentrations. Arthritis Rheum. 2010, 62, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Sulli, A.; Pizzorni, C.; Seriolo, B.; Straub, R.H. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann. Rheum. Dis. 2001, 60, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Corciulo, C.; Cronstein, B.N. Signaling of the Purinergic System in the Joint. Front. Pharmacol. 2019, 10, 1591. [Google Scholar] [CrossRef]
- Blits, M.; Jansen, G.; Assaraf, Y.G.; van de Wiel, M.A.; Lems, W.F.; Nurmohamed, M.T.; van Schaardenburg, D.; Voskuyl, A.E.; Wolbink, G.J.; Vosslamber, S.; et al. Methotrexate normalizes up-regulated folate pathway genes in rheumatoid arthritis. Arthritis Rheum. 2013, 65, 2791–2802. [Google Scholar] [CrossRef]
- Municio, C.; Soler, P.B.; Estrada-Capetillo, L.; Benguria, A.; Dopazo, A.; Garcia-Lorenzo, E.; Fernandez-Arroyo, S.; Joven, J.; Miranda-Carus, M.E.; Gonzalez-Alvaro, I.; et al. Methotrexate selectively targets human proinflammatory macrophages through a thymidylate synthase/p53 axis. Ann. Rheum. Dis. 2016, 75, 2157–2165. [Google Scholar] [CrossRef]
- Van der Heijden, J.W.; Assaraf, Y.G.; Gerards, A.H.; Oerlemans, R.; Lems, W.F.; Scheper, R.J.; Dijkmans, B.A.; Jansen, G. Methotrexate analogues display enhanced inhibition of TNF-alpha production in whole blood from RA patients. Scand. J. Rheumatol. 2014, 43, 9–16. [Google Scholar] [CrossRef]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef]
- Gonen, N.; Assaraf, Y.G. Antifolates in cancer therapy: Structure, activity and mechanisms of drug resistance. Drug Resist. Updat. 2012, 15, 183–210. [Google Scholar] [CrossRef]
- Raz, S.; Stark, M.; Assaraf, Y.G. Folylpoly-gamma-glutamate synthetase: A key determinant of folate homeostasis and antifolate resistance in cancer. Drug Resist. Updat. 2016, 28, 43–64. [Google Scholar] [CrossRef]
- Stark, M.; Raz, S.; Assaraf, Y.G. Folylpolyglutamate synthetase association to the cytoskeleton: Implications to folate metabolon compartmentalization. J. Proteom. 2021, 239, 104169. [Google Scholar] [CrossRef]
- Rots, M.G.; Pieters, R.; Peters, G.J.; Noordhuis, P.; Van Zantwijk, C.H.; Kaspers, G.J.; Hahlen, K.; Creutzig, U.; Veerman, A.J.; Jansen, G. Role of folylpolyglutamate synthetase and folylpolyglutamate hydrolase in methotrexate accumulation and polyglutamylation in childhood leukemia. Blood 1999, 93, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Liani, E.; Rothem, L.; Bunni, M.A.; Smith, C.A.; Jansen, G.; Assaraf, Y.G. Loss of folylpoly-gamma-glutamate synthetase activity is a dominant mechanism of resistance to polyglutamylation-dependent novel antifolates in multiple human leukemia sublines. Int. J. Cancer 2003, 103, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Mauritz, R.; Peters, G.J.; Priest, D.G.; Assaraf, Y.G.; Drori, S.; Kathmann, I.; Noordhuis, P.; Bunni, M.A.; Rosowsky, A.; Schornagel, J.H.; et al. Multiple mechanisms of resistance to methotrexate and novel antifolates in human CCRF-CEM leukemia cells and their implications for folate homeostasis. Biochem. Pharmacol. 2002, 63, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shikano, K.; Nanki, T.; Kawai, S. Folylpolyglutamate synthase is a major determinant of intracellular methotrexate polyglutamates in patients with rheumatoid arthritis. Sci. Rep. 2016, 6, 35615. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.B.; Lin, M.; Struys, E.A.; Heydari, P.; Hebing, R.C.F.; Nurmohamed, M.T.; Van der Laken, C.; Lems, W.F.; Cloos, J.; Jansen, G.; et al. Development and Validation of a Sensitive UHPLC-MS/MS-Based Method for the Analysis of Folylpolyglutamate Synthetase Enzymatic Activity in Peripheral Blood Mononuclear Cells: Application in Rheumatoid Arthritis and Leukemia Patients. Ther. Drug Monit. 2019, 41, 598–606. [Google Scholar] [CrossRef]
- Barredo, J.; Moran, R.G. Determinants of antifolate cytotoxicity: Folylpolyglutamate synthetase activity during cellular proliferation and development. Mol. Pharmacol. 1992, 42, 687–694. [Google Scholar]
- Stark, M.; Wichman, C.; Avivi, I.; Assaraf, Y.G. Aberrant splicing of folylpolyglutamate synthetase as a novel mechanism of antifolate resistance in leukemia. Blood 2009, 113, 4362–4369. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Raz, S.; Stark, M.; Assaraf, Y.G.; Jansen, G.; Peters, G.J.; Sonneveld, E.; Kaspers, G.J.; Cloos, J. Folylpolyglutamate synthetase splicing alterations in acute lymphoblastic leukemia are provoked by methotrexate and other chemotherapeutics and mediate chemoresistance. Int. J. Cancer 2016, 138, 1645–1656. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updat. 2020, 53, 100728. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Assaraf, Y.G.; Hoekstra, M.; Sciarrillo, R.; Jansen, G.; Peters, G.J.; Pieters, R.; Sonneveld, E.; Escherich, G.; Kaspers, G.J.; et al. The association of aberrant folylpolyglutamate synthetase splicing with ex vivo methotrexate resistance and clinical outcome in childhood acute lymphoblastic leukemia. Haematologica 2016, 101, e291–e294. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.B.; Lin, M.; Lems, W.F.; Ter Wee, M.M.; Wojtuszkiewicz, A.; Nurmohamed, M.T.; Cloos, J.; Assaraf, Y.G.; Jansen, G.; De Jonge, R. Association of altered folylpolyglutamate synthetase pre-mRNA splicing with methotrexate unresponsiveness in early rheumatoid arthritis. Rheumatology 2021, 60, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef]
- Nakashima-Matsushita, N.; Homma, T.; Yu, S.; Matsuda, T.; Sunahara, N.; Nakamura, T.; Tsukano, M.; Ratnam, M.; Matsuyama, T. Selective expression of folate receptor beta and its possible role in methotrexate transport in synovial macrophages from patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1609–1616. [Google Scholar] [CrossRef]
- Fuentelsaz-Romero, S.; Barrio-Alonso, C.; Garcia Campos, R.; Torres Torresano, M.; Muller, I.B.; Triguero-Martinez, A.; Nuno, L.; Villalba, A.; Garcia-Vicuna, R.; Jansen, G.; et al. The Macrophage Reprogramming Ability of Antifolates Reveals Soluble CD14 as a Potential Biomarker for Methotrexate Response in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 776879. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, R.; Blits, M.; Dijkmans, B.A.C.; Van der Heijden, J.W.; Lems, W.F.; Scheffer, G.L.; Van de Ven, R.; Peters, G.J.; Assaraf, Y.G.; Scheper, R.J.; et al. Expression profiling of ABC transporters in peripheral blood lymphocytes and monocyte-derived macrophages of rheumatoid arthritis patients. J. Mol. Clin. Med. 2020, 3, 47–60. [Google Scholar] [CrossRef]
- Van der Heijden, J.W.; Oerlemans, R.; Dijkmans, B.A.; Qi, H.; Van der Laken, C.J.; Lems, W.F.; Jackman, A.L.; Kraan, M.C.; Tak, P.P.; Ratnam, M.; et al. Folate receptor beta as a potential delivery route for novel folate antagonists to macrophages in the synovial tissue of rheumatoid arthritis patients. Arthritis Rheum. 2009, 60, 12–21. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Tak, P.P. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009, 60, 1210–1221. [Google Scholar] [CrossRef]
- Udalova, I.A.; Mantovani, A.; Feldmann, M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 472–485. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, M.; Jia, S. Macrophage: Key player in the pathogenesis of autoimmune diseases. Front. Immunol. 2023, 14, 1080310. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, R.; Oerlemans, R.; Van der Heijden, J.W.; Scheffer, G.L.; De Gruijl, T.D.; Jansen, G.; Scheper, R.J. ABC drug transporters and immunity: Novel therapeutic targets in autoimmunity and cancer. J. Leukoc. Biol. 2009, 86, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Hooijberg, J.H.; Broxterman, H.J.; Kool, M.; Assaraf, Y.G.; Peters, G.J.; Noordhuis, P.; Scheper, R.J.; Borst, P.; Pinedo, H.M.; Jansen, G. Antifolate resistance mediated by the multidrug resistance proteins MRP1 and MRP2. Cancer Res. 1999, 59, 2532–2535. [Google Scholar] [PubMed]
- Assaraf, Y.G.; Rothem, L.; Hooijberg, J.H.; Stark, M.; Ifergan, I.; Kathmann, G.A.; Dijkmans, B.A.; Peters, G.J.; Jansen, G. Loss of multidrug resistance protein 1 (MRP1) expression and folate efflux activity results in a highly concentrative folate transport in human leukemia cells. J. Biol. Chem. 2003, 278, 6680–6686. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.; Rothem, L.; Jansen, G.; Scheffer, G.L.; Goldman, I.D.; Assaraf, Y.G. Antifolate Resistance Associated with Loss of MRP1 Expression and Function in Chinese Hamster Ovary Cells with Markedly Impaired Export of Folate and Cholate. Mol. Pharmacol. 2003, 64, 220–227. [Google Scholar] [CrossRef]
- Ifergan, I.; Shafran, A.; Jansen, G.; Hooijberg, J.H.; Scheffer, G.L.; Assaraf, Y.G. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression. A role for BCRP in cellular folate homeostasis. J. Biol. Chem. 2004, 279, 25527–25534. [Google Scholar] [CrossRef]
- Chara, L.; Sanchez-Atrio, A.; Perez, A.; Cuende, E.; Albarran, F.; Turrion, A.; Chevarria, J.; Del Barco, A.A.; Sanchez, M.A.; Monserrat, J.; et al. The number of circulating monocytes as biomarkers of the clinical response to methotrexate in untreated patients with rheumatoid arthritis. J. Transl. Med. 2015, 13, 2. [Google Scholar] [CrossRef]
- Zhao, R.; Matherly, L.H.; Goldman, I.D. Membrane transporters and folate homeostasis: Intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med. 2009, 11, e4. [Google Scholar] [CrossRef]
- Lasry, I.; Berman, B.; Straussberg, R.; Sofer, Y.; Bessler, H.; Sharkia, M.; Glaser, F.; Jansen, G.; Drori, S.; Assaraf, Y.G. A novel loss-of-function mutation in the proton-coupled folate transporter from a patient with hereditary folate malabsorption reveals that Arg 113 is crucial for function. Blood 2008, 112, 2055–2061. [Google Scholar] [CrossRef]
- Lasry, I.; Berman, B.; Glaser, F.; Jansen, G.; Assaraf, Y.G. Hereditary folate malabsorption: A positively charged amino acid at position 113 of the proton-coupled folate transporter (PCFT/SLC46A1) is required for folic acid binding. Biochem. Biophys. Res. Commun. 2009, 386, 426–431. [Google Scholar] [CrossRef]
- Frigerio, B.; Bizzoni, C.; Jansen, G.; Leamon, C.P.; Peters, G.J.; Low, P.S.; Matherly, L.H.; Figini, M. Folate receptors and transporters: Biological role and diagnostic/therapeutic targets in cancer and other diseases. J. Exp. Clin. Cancer Res. 2019, 38, 125. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G. The role of multidrug resistance efflux transporters in antifolate resistance and folate homeostasis. Drug Resist. Updat. 2006, 9, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, J.W.; Oerlemans, R.; Tak, P.P.; Assaraf, Y.G.; Kraan, M.C.; Scheffer, G.L.; Van der Laken, C.J.; Lems, W.F.; Scheper, R.J.; Dijkmans, B.A.; et al. Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis Rheum. 2009, 60, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Shafran, A.; Ifergan, I.; Bram, E.; Jansen, G.; Kathmann, I.; Peters, G.J.; Robey, R.W.; Bates, S.E.; Assaraf, Y.G. ABCG2 harboring the Gly482 mutation confers high-level resistance to various hydrophilic antifolates. Cancer Res. 2005, 65, 8414–8422. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, Z.; Zhang, K.; Chi, Z.; Xu, T.; Jiang, D.; Chen, S.; Li, W.; Yang, X.; Zhang, X.; et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 2019, 75, 1147–1160. [Google Scholar] [CrossRef]
- Municio, C.; Dominguez-Soto, A.; Fuentelsaz-Romero, S.; Lamana, A.; Montes, N.; Cuevas, V.D.; Campos, R.G.; Pablos, J.L.; Gonzalez-Alvaro, I.; Puig-Kroger, A. Methotrexate limits inflammation through an A20-dependent cross-tolerance mechanism. Ann. Rheum. Dis. 2018, 77, 752–759. [Google Scholar] [CrossRef]
- Soler, P.B.; Estrada-Capetillo, L.; Izquierdo, E.; Criado, G.; Nieto, C.; Municio, C.; Gonzalez-Alvaro, I.; Sanchez-Mateos, P.; Pablos, J.L.; Corbi, A.L.; et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J. Pathol. 2015, 235, 515–526. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef]
- Peres, R.S.; Liew, F.Y.; Talbot, J.; Carregaro, V.; Oliveira, R.D.; Almeida, S.L.; Franca, R.F.; Donate, P.B.; Pinto, L.G.; Ferreira, F.I.; et al. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 2509–2514. [Google Scholar] [CrossRef]
- Chandrupatla, D.M.S.H.; Molthoff, C.F.M.; Ritsema, W.I.G.R.; Vos, R.; Elshof, E.; Matsuyama, T.; Low, P.S.; Musters, R.J.P.; Hammond, A.; Windhorst, A.D.; et al. Prophylactic and therapeutic activity of alkaline phosphatase in arthritic rats: Single-agent effects of alkaline phosphatase and synergistic effects in combination with methotrexate. Transl. Res. 2018, 199, 24–38. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Van den Bossche, J. Immunometabolism in the Single-Cell Era. Cell Metab. 2020, 32, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wu, B.; Goodman, S.B.; Berry, G.J.; Goronzy, J.J.; Weyand, C.M. Metabolic Control of Autoimmunity and Tissue Inflammation in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 652771. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Harber, K.J.; De Goede, K.E.; Verberk, S.G.S.; Meinster, E.; De Vries, H.E.; Van Weeghel, M.; De Winther, M.P.J.; Van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef]
- Saraiva, A.L.; Veras, F.P.; Peres, R.S.; Talbot, J.; de Lima, K.A.; Luiz, J.P.; Carballido, J.M.; Cunha, T.M.; Cunha, F.Q.; Ryffel, B.; et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of Th17 cells in the lymph nodes. FASEB J. 2018, 32, 6550–6558. [Google Scholar] [CrossRef]
- Daly, R.; Blackburn, G.; Best, C.; Goodyear, C.S.; Mudaliar, M.; Burgess, K.; Stirling, A.; Porter, D.; McInnes, I.B.; Barrett, M.P.; et al. Changes in Plasma Itaconate Elevation in Early Rheumatoid Arthritis Patients Elucidates Disease Activity Associated Macrophage Activation. Metabolites 2020, 10, 241. [Google Scholar] [CrossRef]
- Gosselt, H.R.; Muller, I.B.; Jansen, G.; van Weeghel, M.; Vaz, F.M.; Hazes, J.M.W.; Heil, S.G.; de Jonge, R. Identification of Metabolic Biomarkers in Relation to Methotrexate Response in Early Rheumatoid Arthritis. J. Pers. Med. 2020, 10, 271. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Hou, Z.; Gangjee, A.; Matherly, L.H. Therapeutic Targeting of Mitochondrial One-Carbon Metabolism in Cancer. Mol. Cancer Ther. 2020, 19, 2245–2255. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lin, T.Y.; Lee, G.; Paddock, M.N.; Momb, J.; Cheng, Z.; Li, Q.; Fei, D.L.; Stein, B.D.; Ramsamooj, S.; et al. Mitochondrial One-Carbon Pathway Supports Cytosolic Folate Integrity in Cancer Cells. Cell 2018, 175, 1546–1560. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1beta Production. Cell Metab. 2019, 29, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, S.A.; Hackett, J.C.; Moran, R.G. Tetrahydrofolate recognition by the mitochondrial folate transporter. J. Biol. Chem. 2011, 286, 31480–31489. [Google Scholar] [CrossRef]
- Lawrence, S.A.; Titus, S.A.; Ferguson, J.; Heineman, A.L.; Taylor, S.M.; Moran, R.G. Mammalian mitochondrial and cytosolic folylpolyglutamate synthetase maintain the subcellular compartmentalization of folates. J. Biol. Chem. 2014, 289, 29386–29396. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Ning, C.; Nayeen, M.J.; Shah, K.; Kalpage, H.; Fruhauf, J.; Wallace-Povirk, A.; O’Connor, C.; Hou, Z.; Kim, S.; et al. Cellular Pharmacodynamics of a Novel Pyrrolo[3,2-d]pyrimidine Inhibitor Targeting Mitochondrial and Cytosolic One-Carbon Metabolism. Mol. Pharmacol. 2020, 97, 9–22. [Google Scholar] [CrossRef]
- De Rotte, M.C.; De Jong, P.H.; Pluijm, S.M.; Calasan, M.B.; Barendregt, P.J.; Van Zeben, D.; Van der Lubbe, P.A.; De Sonnaville, P.B.; Lindemans, J.; Hazes, J.M.; et al. Association of low baseline levels of erythrocyte folate with treatment nonresponse at three months in rheumatoid arthritis patients receiving methotrexate. Arthritis Rheum. 2013, 65, 2803–2813. [Google Scholar] [CrossRef]
- Puig-Kroger, A.; Sierra-Filardi, E.; Dominguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gomez-Aguado, F.; Ratnam, M.; Sanchez-Mateos, P.; Corbi, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Kooi, I.E.; Leon, L.G.; Sonneveld, E.; Kuiper, R.P.; Jansen, G.; Giovannetti, E.; Kaspers, G.J.L.; Cloos, J. Glucocorticoid Resistant Pediatric Acute Lymphoblastic Leukemia Samples Display Altered Splicing Profile and Vulnerability to Spliceosome Modulation. Cancers 2020, 12, 723. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.B.; Meijers, S.; Kampstra, P.; van Dijk, S.; van Elswijk, M.; Lin, M.; Wojtuszkiewicz, A.M.; Jansen, G.; de Jonge, R.; Cloos, J. Computational comparison of common event-based differential splicing tools: Practical considerations for laboratory researchers. BMC Bioinformatics 2021, 22, 347. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Driehuis, E.; Oosterom, N.; Heil, S.G.; Muller, I.B.; Lin, M.; Kolders, S.; Jansen, G.; de Jonge, R.; Pieters, R.; Clevers, H.; et al. Patient-derived oral mucosa organoids as an in vitro model for methotrexate induced toxicity in pediatric acute lymphoblastic leukemia. PLoS ONE 2020, 15, e0231588. [Google Scholar] [CrossRef]
- Hebing, R.C.; Lin, M.; Bulatovic-Calasan, M.; Muller, I.B.; Mahmoud, S.; Heil, S.; Struys, E.A.; Van den Bemt, B.J.; Twisk, J.W.; Lems, W.; et al. Pharmacokinetics of oral and subcutaneous methotrexate in red and white blood cells in patients with early rheumatoid arthritis: The methotrexate monitoring trial. Ann. Rheum. Dis. 2023, 82, 460–467. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muller, I.B.; Lin, M.; Jonge, R.d.; Will, N.; López-Navarro, B.; Laken, C.v.d.; Struys, E.A.; Oudejans, C.B.M.; Assaraf, Y.G.; Cloos, J.; et al. Methotrexate Provokes Disparate Folate Metabolism Gene Expression and Alternative Splicing in Ex Vivo Monocytes and GM-CSF- and M-CSF-Polarized Macrophages. Int. J. Mol. Sci. 2023, 24, 9641. https://doi.org/10.3390/ijms24119641
Muller IB, Lin M, Jonge Rd, Will N, López-Navarro B, Laken Cvd, Struys EA, Oudejans CBM, Assaraf YG, Cloos J, et al. Methotrexate Provokes Disparate Folate Metabolism Gene Expression and Alternative Splicing in Ex Vivo Monocytes and GM-CSF- and M-CSF-Polarized Macrophages. International Journal of Molecular Sciences. 2023; 24(11):9641. https://doi.org/10.3390/ijms24119641
Chicago/Turabian StyleMuller, Ittai B., Marry Lin, Robert de Jonge, Nico Will, Baltasar López-Navarro, Conny van der Laken, Eduard A. Struys, Cees B. M. Oudejans, Yehuda G. Assaraf, Jacqueline Cloos, and et al. 2023. "Methotrexate Provokes Disparate Folate Metabolism Gene Expression and Alternative Splicing in Ex Vivo Monocytes and GM-CSF- and M-CSF-Polarized Macrophages" International Journal of Molecular Sciences 24, no. 11: 9641. https://doi.org/10.3390/ijms24119641
APA StyleMuller, I. B., Lin, M., Jonge, R. d., Will, N., López-Navarro, B., Laken, C. v. d., Struys, E. A., Oudejans, C. B. M., Assaraf, Y. G., Cloos, J., Puig-Kröger, A., & Jansen, G. (2023). Methotrexate Provokes Disparate Folate Metabolism Gene Expression and Alternative Splicing in Ex Vivo Monocytes and GM-CSF- and M-CSF-Polarized Macrophages. International Journal of Molecular Sciences, 24(11), 9641. https://doi.org/10.3390/ijms24119641