


Bioinformatics Analysis of Gene Expression Profiles for Diagnosing Sepsis and Risk Prediction in Patients with Sepsis



Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics of Patients with Sepsis
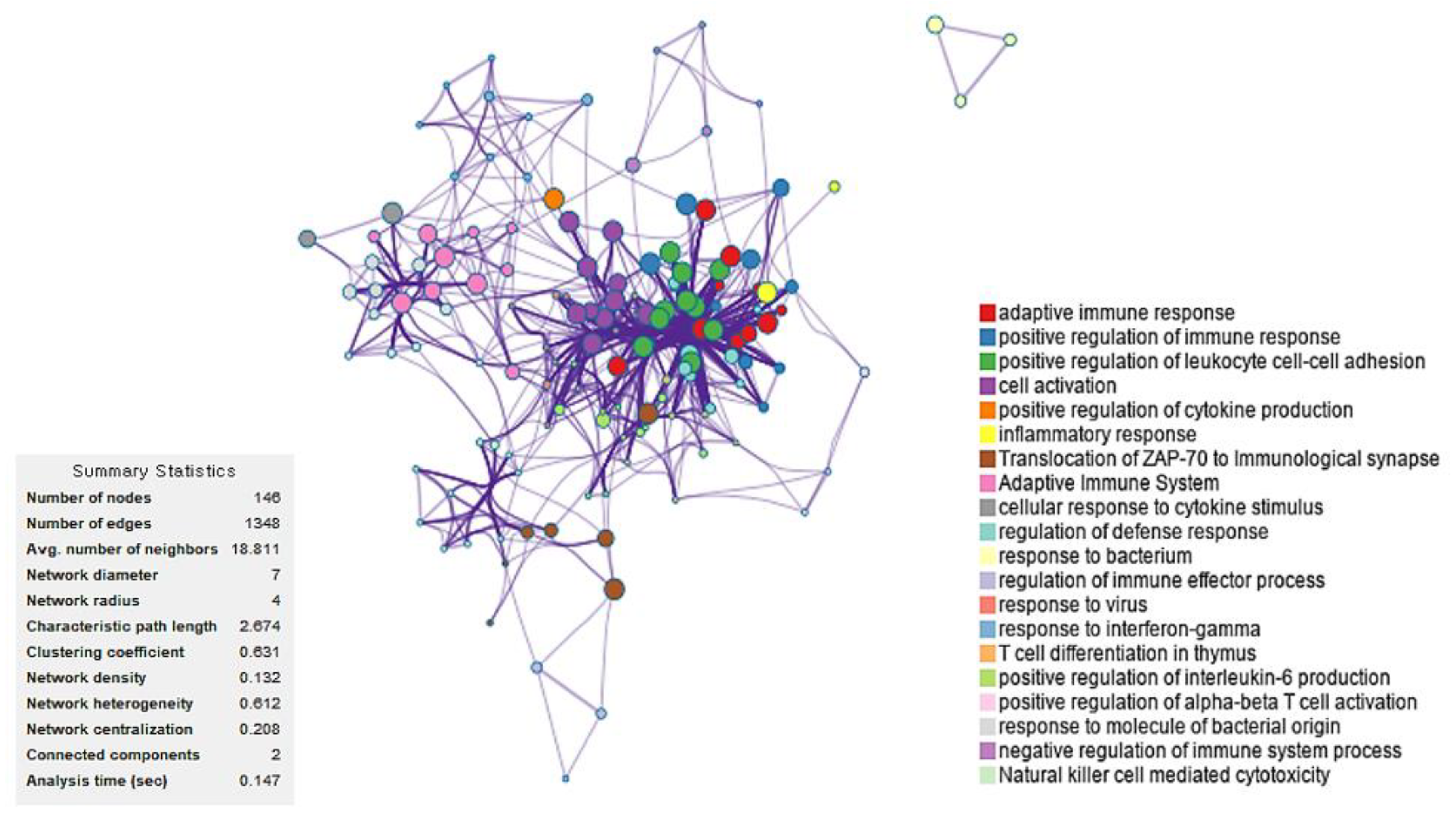
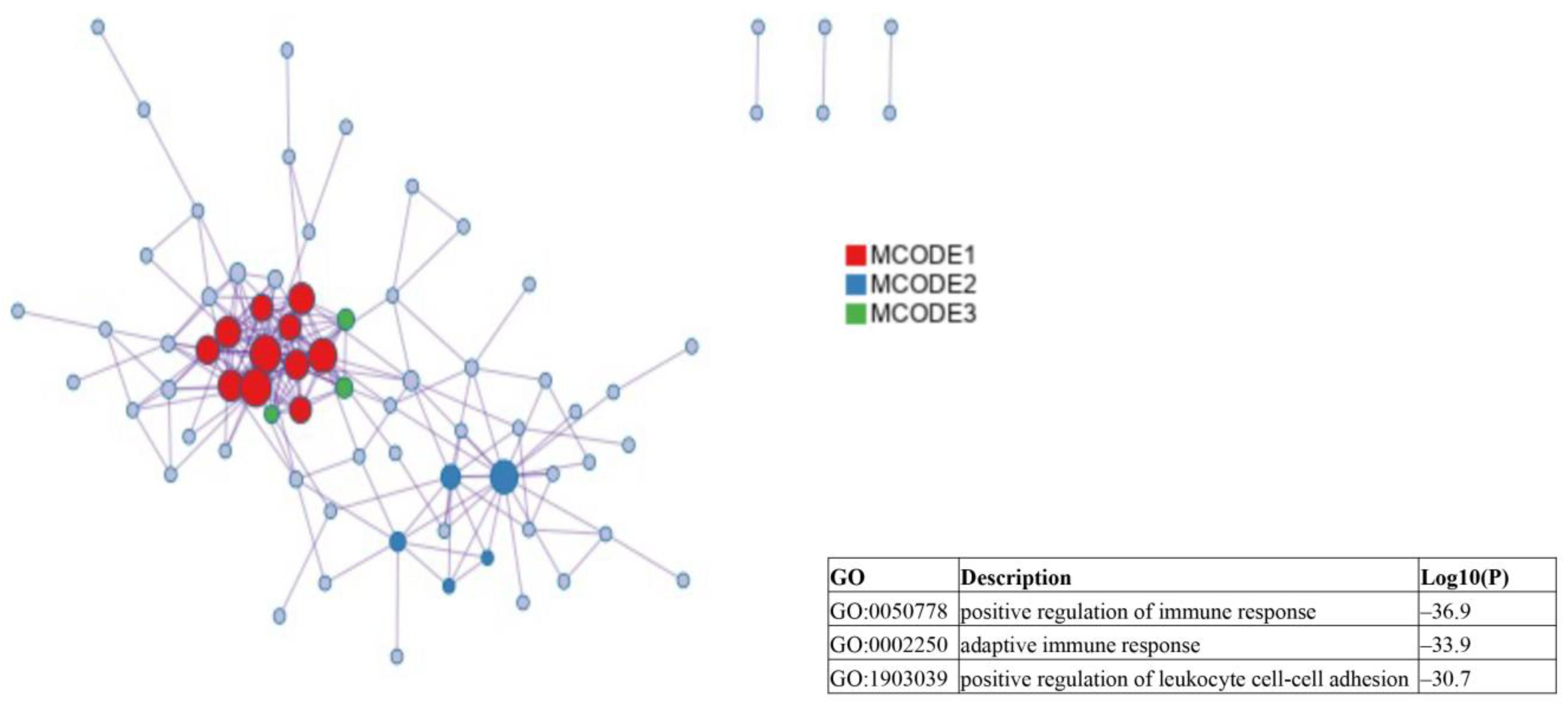
2.2. Identification of Candidate mRNAs: Bioinformatics Analyses
2.3. Experimental and Clinical Validation of Potential Biomarkers from the mRNA Profile
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Sample Collection
4.3. Total RNA Isolation and Quality Analysis
4.4. Library Preparation and Sequencing
4.5. Data Analysis
4.6. Identification of DEGs
4.7. Functional and Pathway Enrichment Analyses
4.8. Protein–Protein Interaction Network Analysis
4.9. Quantitative Real-Time PCR
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K.; International Forum of Acute Care Trialists. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann-Struzek, C.; Mellhammar, L.; Rose, N.; Cassini, A.; Rudd, K.E.; Schlattmann, P.; Allegranzi, B.; Reinhart, K. Incidence and mortality of hospital- and ICU-treated sepsis: Results from an updated and expanded systematic review and meta-analysis. Intensive Care Med. 2020, 46, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Gaieski, D.F.; Edwards, J.M.; Kallan, M.J.; Carr, B.G. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit. Care Med. 2013, 41, 1167–1174. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Cooke, C.R.; Wunsch, H.; Kahn, J.M. Population burden of long-term survivorship after severe sepsis in older Americans. J. Am. Geriatr. Soc. 2012, 60, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Gesten, F.; Prescott, H.C.; Friedrich, M.E.; Iwashyna, T.J.; Phillips, G.S.; Lemeshow, S.; Osborn, T.; Terry, K.M.; Levy, M.M. Time to Treatment and Mortality during Mandated Emergency Care for Sepsis. N. Engl. J. Med. 2017, 376, 2235–2244. [Google Scholar] [CrossRef]
- Simon, L.; Saint-Louis, P.; Amre, D.K.; Lacroix, J.; Gauvin, F. Procalcitonin and C-reactive protein as markers of bacterial infection in critically ill children at onset of systemic inflammatory response syndrome. Pediatr. Crit. Care Med. 2008, 9, 407–413. [Google Scholar] [CrossRef]
- Yang, Y.; Xie, J.; Guo, F.; Longhini, F.; Gao, Z.; Huang, Y.; Qiu, H. Combination of C-reactive protein, procalcitonin and sepsis-related organ failure score for the diagnosis of sepsis in critical patients. Ann. Intensive Care 2016, 6, 51. [Google Scholar] [CrossRef]
- Pierrakos, C.; Vincent, J.L. Sepsis biomarkers: A review. Crit. Care 2010, 14, R15. [Google Scholar] [CrossRef]
- Yoo, H.; Lee, J.Y.; Park, J.; Yang, J.H.; Suh, G.Y.; Jeon, K. Association of Plasma Level of TNF-Related Apoptosis-Inducing Ligand with Severity and Outcome of Sepsis. J. Clin. Med. 2020, 9, 1661. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, L.; Zhong, W.; Chen, M.; Zhang, Q. Bioinformatics Analysis of Gene Expression Profiles for Risk Prediction in Patients with Septic Shock. Med. Sci. Monit. 2019, 25, 9563–9571. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, B.; Yi, W.; Wang, F.; Wei, S.; Yan, H.; Qin, P.; Zou, D.; Wei, R.; Chen, N. Identification of Potential Early Diagnostic Biomarkers of Sepsis. J. Inflamm. Res. 2021, 14, 621–631. [Google Scholar] [CrossRef]
- Niu, J.; Qin, B.; Wang, C.; Chen, C.; Yang, J.; Shao, H. Identification of Key Immune-Related Genes in the Progression of Septic Shock. Front. Genet. 2021, 12, 668527. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Tan, L.; Zhou, Y.; Zhu, Y.; Ma, C.; Wu, Y.; Du, Y.; Liu, L.; Hu, Y.; Mao, Q.; et al. The Landscape of Featured Metabolism-Related Genes and Imbalanced Immune Cell Subsets in Sepsis. Front. Genet. 2022, 13, 821275. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Feng, J.; Yang, Y.; Zhao, R.; Yu, Q.; Qin, H.; Wei, L.; Ji, P.; Li, H.; Wu, Z.; et al. Screening of Key Genes of Sepsis and Septic Shock Using Bioinformatics Analysis. J. Inflamm. Res. 2021, 14, 829–841. [Google Scholar] [CrossRef]
- Wu, T.; Liang, X.; Jiang, Y.; Chen, Q.; Zhang, H.; Zhang, S.; Zhang, C.; Lv, Y.; Xin, J.; Jiang, J.; et al. Comprehensive Transcriptome Profiling of Peripheral Blood Mononuclear Cells from Patients with Sepsis. Int. J. Med. Sci. 2020, 17, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Herwanto, V.; Tang, B.; Wang, Y.; Shojaei, M.; Nalos, M.; Shetty, A.; Lai, K.; McLean, A.S.; Schughart, K. Blood transcriptome analysis of patients with uncomplicated bacterial infection and sepsis. BMC Res. Notes 2021, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Qi, A.; Zhang, Y.; Jiao, L.; Liu, Y.; Shou, S. Bioinformatics Analysis for Multiple Gene Expression Profiles in Sepsis. Med. Sci. Monit. 2020, 26, e920818. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.L.; Chen, H.H.; Jiang, Y.H.; Hsiao, T.H.; Wang, C.Y.; Wu, C.L.; Ko, T.M.; Chao, W.C. Using RNA-Seq to Investigate Immune-Metabolism Features in Immunocompromised Patients With Sepsis. Front. Med. 2021, 8, 747263. [Google Scholar] [CrossRef]
- National Library of Medicine (US), N.C.f.B.I. Gene. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 31 January 2023).
- Ulas, T.; Pirr, S.; Fehlhaber, B.; Bickes, M.S.; Loof, T.G.; Vogl, T.; Mellinger, L.; Heinemann, A.S.; Burgmann, J.; Schöning, J.; et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat. Immunol. 2017, 18, 622–632. [Google Scholar] [CrossRef]
- Boyd, J.H.; Kan, B.; Roberts, H.; Wang, Y.; Walley, K.R. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ. Res. 2008, 102, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Forel, J.M.; Chiche, L.; Thomas, G.; Mancini, J.; Farnarier, C.; Cognet, C.; Guervilly, C.; Daumas, A.; Vély, F.; Xéridat, F.; et al. Phenotype and functions of natural killer cells in critically-ill septic patients. PLoS ONE 2012, 7, e50446. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, Y.; Cui, Y.; Ren, Y.; Li, R.; Rong, Q. Inhibition of microRNA-155 relieves sepsis-induced liver injury through inactivating the JAK/STAT pathway. Mol. Med. Rep. 2015, 12, 6013–6018. [Google Scholar] [CrossRef]
- Winkler, M.S.; Rissiek, A.; Priefler, M.; Schwedhelm, E.; Robbe, L.; Bauer, A.; Zahrte, C.; Zoellner, C.; Kluge, S.; Nierhaus, A. Human leucocyte antigen (HLA-DR) gene expression is reduced in sepsis and correlates with impaired TNFα response: A diagnostic tool for immunosuppression? PLoS ONE 2017, 12, e0182427. [Google Scholar] [CrossRef]
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol. 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Yoo, H.; Lee, J.Y.; Park, J.; Jeon, K. Plasma Mitochondrial DNA and Necroptosis as Prognostic Indicators in Critically Ill Patients with Sepsis. Biomedicines 2022, 10, 2386. [Google Scholar] [CrossRef]
- Li, D.; Ren, W.; Wang, X.; Wang, F.; Gao, Y.; Ning, Q.; Han, Y.; Song, T.; Lu, S. A modified method using TRIzol reagent and liquid nitrogen produces high-quality RNA from rat pancreas. Appl. Biochem. Biotechnol. 2009, 158, 253–261. [Google Scholar] [CrossRef]
- Sp, N.; Kang, D.Y.; Jo, E.S.; Rugamba, A.; Kim, W.S.; Park, Y.M.; Hwang, D.Y.; Yoo, J.S.; Liu, Q.; Jang, K.J.; et al. Tannic Acid Promotes TRAIL-Induced Extrinsic Apoptosis by Regulating Mitochondrial ROS in Human Embryonic Carcinoma Cells. Cells 2020, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc 2015, 2015, pdb-top084970. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge–Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://www.R-project.org (accessed on 5 January 2023).
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef]
- Udhaya Kumar, S.; Thirumal Kumar, D.; Bithia, R.; Sankar, S.; Magesh, R.; Sidenna, M.; George Priya Doss, C.; Zayed, H. Analysis of Differentially Expressed Genes and Molecular Pathways in Familial Hypercholesterolemia Involved in Atherosclerosis: A Systematic and Bioinformatics Approach. Front. Genet. 2020, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Wang, J.; Zhong, J.; Chen, G.; Li, M.; Wu, F.X.; Pan, Y. ClusterViz: A Cytoscape APP for Cluster Analysis of Biological Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Maru, D.M.; Singh, R.R.; Hannah, C.; Albarracin, C.T.; Li, Y.X.; Abraham, R.; Romans, A.M.; Yao, H.; Luthra, M.G.; Anandasabapathy, S.; et al. MicroRNA-196a is a potential marker of progression during Barrett’s metaplasia-dysplasia-invasive adenocarcinoma sequence in esophagus. Am. J. Pathol. 2009, 174, 1940–1948. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Value (n = 133) |
|---|---|
| Age (years) | 66 (58–73) |
| Male | 90 (67.7) |
| Comorbidities | |
| All malignancies | 51 (38.3) |
| Solid organ malignancies | 35 (26.3) |
| Hematologic malignancies | 16 (12.0) |
| Diabetes mellitus | 41 (30.8) |
| Chronic obstructive pulmonary disease | 16 (12.0) |
| Chronic kidney disease | 10 (7.5) |
| Myocardial infarction | 8 (6.0) |
| Congestive heart failure | 7 (5.3) |
| Cerebrovascular disease | 8 (6.0) |
| Chronic liver disease | 11 (8.3) |
| Charlson Comorbidity Index | 2 (1–3) |
| Clinical status on ICU admission | |
| Mechanical ventilation | 60 (45.1) |
| Vasopressor support | 69 (51.9) |
| Laboratory findings | |
| WBC (/µL) | 13,640 (5060–20,340) |
| Hemoglobin (g/dL) | 10.1 (8.9–11.7) |
| Platelet (/µL) | 136,000 (59,000–214,000) |
| Albumin (g/dL) | 2.9 (2.6–3.2) |
| CRP (mg/dL) | 12.5 (5.8–24.5) |
| Lactate (mg/dL) | 3.0 (2.0–4.4) |
| PaO2/FiO2 ratio | 216 (135–323) |
| Severity of illness | |
| SAPS 3 score | 55 (47–63) |
| APACHE II score | 24 (20–30) |
| SOFA score, initial | 9 (7–11) |
| Outcome | |
| 28-day mortality | 22 (16.5) |
| In-hospital mortality | 33 (24.8) |
| Top 10 Upregulated Genes | Top 10 Downregulated Genes | ||||
|---|---|---|---|---|---|
| Entrez ID | Gene Symbol | Fold Change | Entrez ID | Gene Symbol | Fold Change |
| 6279 | S100A8 | 16.17 | 973 | CD79A | 0.12 |
| 8876 | VNN1 | 7.83 | 3120 | HLA-DQB2 | 0.13 |
| 3148 | HMGB2 | 7.23 | 122618 | PLD4 | 0.14 |
| 6280 | S100A9 | 5.43 | 1236 | CCR7 | 0.15 |
| 301 | ANXA1 | 5.42 | 259197 | NCR3 | 0.17 |
| 665 | BNIP3L | 4.67 | 29802 | VPREB3 | 0.18 |
| 200315 | APOBEC3A | 4.61 | 6932 | TCF7 | 0.199 |
| 353514 | LILRA5 | 4.49 | 933 | CD22 | 0.201 |
| 1378 | CR1 | 4.46 | 10578 | GNLY | 0.206 |
| 1604 | CD55 | 4.29 | 974 | CD79B | 0.213 |
| Patients with Sepsis vs. Healthy Volunteers * | ||||
|---|---|---|---|---|
| Patients with Sepsis | Healthy Volunteers | p-Value | ||
| Upregulated genes | S100A8 | 19.8 (11.6–24.2) | 7.6 (7.1–8.2) | <0.001 |
| S100A9 | 7.6 (3.8–10.3) | 3.8 (2.6–4.2) | <0.001 | |
| CR1 | 5.5 (2.5–8.8) | 2.7 (2.3–2.8) | 0.005 | |
| Downregulated genes | CD79A | 0.3 (0.2–0.4) | 0.5 (0.5–0.6) | <0.001 |
| HLA-DQB2 | 0.2 (0.2–0.2) | 0.3 (0.3–0.4) | <0.001 | |
| PLD4 | 0.5 (0.2–0.7) | 0.9 (0.9–1.0) | <0.001 | |
| CCR7 | 0.3 (0.1–0.4) | 0.5 (0.5–0.6) | <0.001 | |
| The Dead vs. Surviving among Patients with Sepsis * | ||||
| Patients (Dead) | Patients (Surviving) | p-Value | ||
| Upregulated genes | S100A8 | 25.9 (24.1–27.8) | 17.1 (11.4–21.2) | <0.001 |
| S100A9 | 10.7 (9.7–11.8) | 6.3 (3.2–8.5) | <0.001 | |
| CR1 | 10.3 (9.3–11.9) | 4.0 (2.0–6.6) | <0.001 | |
| Downregulated genes | CD79A | 0.5 (0.4–0.6) | 0.2 (0.1–0.3) | <0.001 |
| HLA-DQB2 | 0.3 (0.2–0.3) | 0.2 (0.1–0.2) | <0.001 | |
| PLD4 | 0.7 (0.6–0.8) | 0.4 (0.2–0.6) | <0.001 | |
| CCR7 | 0.4 (0.4–0.5) | 0.2 (0.1–0.3) | <0.001 | |
| mRNAs | Sepsis Diagnosis | |||||
|---|---|---|---|---|---|---|
| AUC | SE | 95% CI | Optimal Cut-Off | Sensitivity | Specificity | |
| S100A8 | 0.931 | 0.026 | 0.880–0.982 | ≥9.0 | 94.0% | 83.3% |
| S100A9 | 0.791 | 0.041 | 0.711–0.871 | ≥5.0 | 69.2% | 100% |
| CR1 | 0.747 | 0.037 | 0.674–0.820 | ≥3.0 | 72.9% | 100% |
| CD79A | 0.113 | 0.028 | 0.059–0.168 | NA | NA | NA |
| HLA-DQB2 | 0.003 | 0.003 | 0–0.009 | NA | NA | NA |
| PLD4 | 0.013 | 0.013 | 0–0.038 | NA | NA | NA |
| CCR7 | 0.016 | 0.010 | 0–0.036 | NA | NA | NA |
| Mortality Prediction in Sepsis | ||||||
| AUC | SE | 95% CI | Optimal Cut-Off | Sensitivity | Specificity | |
| S100A8 | 0.919 | 0.026 | 0.868–0.970 | ≥23.6 | 84.9% | 88.0% |
| S100A9 | 0.863 | 0.033 | 0.797–0.928 | ≥9.6 | 78.8% | 85.0% |
| CR1 | 0.966 | 0.014 | 0.939–0.993 | ≥9.0 | 84.9% | 95.0% |
| CD79A | 0.961 | 0.016 | 0.930–0.992 | ≥0.42 | 93.9% | 92.0% |
| HLA-DQB2 | 0.936 | 0.021 | 0.895–0.978 | ≥0.23 | 84.9% | 88.0% |
| PLD4 | 0.918 | 0.026 | 0.866–0.969 | ≥0.64 | 84.9% | 88.0% |
| CCR7 | 0.918 | 0.026 | 0.866–0.969 | ≥0.39 | 78.8% | 90.0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Lee, J.Y.; Yoo, H.; Jeon, K. Bioinformatics Analysis of Gene Expression Profiles for Diagnosing Sepsis and Risk Prediction in Patients with Sepsis. Int. J. Mol. Sci. 2023, 24, 9362. https://doi.org/10.3390/ijms24119362
Choi H, Lee JY, Yoo H, Jeon K. Bioinformatics Analysis of Gene Expression Profiles for Diagnosing Sepsis and Risk Prediction in Patients with Sepsis. International Journal of Molecular Sciences. 2023; 24(11):9362. https://doi.org/10.3390/ijms24119362
Chicago/Turabian StyleChoi, Hayoung, Jin Young Lee, Hongseok Yoo, and Kyeongman Jeon. 2023. "Bioinformatics Analysis of Gene Expression Profiles for Diagnosing Sepsis and Risk Prediction in Patients with Sepsis" International Journal of Molecular Sciences 24, no. 11: 9362. https://doi.org/10.3390/ijms24119362
APA StyleChoi, H., Lee, J. Y., Yoo, H., & Jeon, K. (2023). Bioinformatics Analysis of Gene Expression Profiles for Diagnosing Sepsis and Risk Prediction in Patients with Sepsis. International Journal of Molecular Sciences, 24(11), 9362. https://doi.org/10.3390/ijms24119362