
Genetic Ablation of LAT1 Inhibits Growth of Liver Cancer Cells and Downregulates mTORC1 Signaling

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Expression of LAT1 Is Associated with Clinical Outcomes of HCC Patients
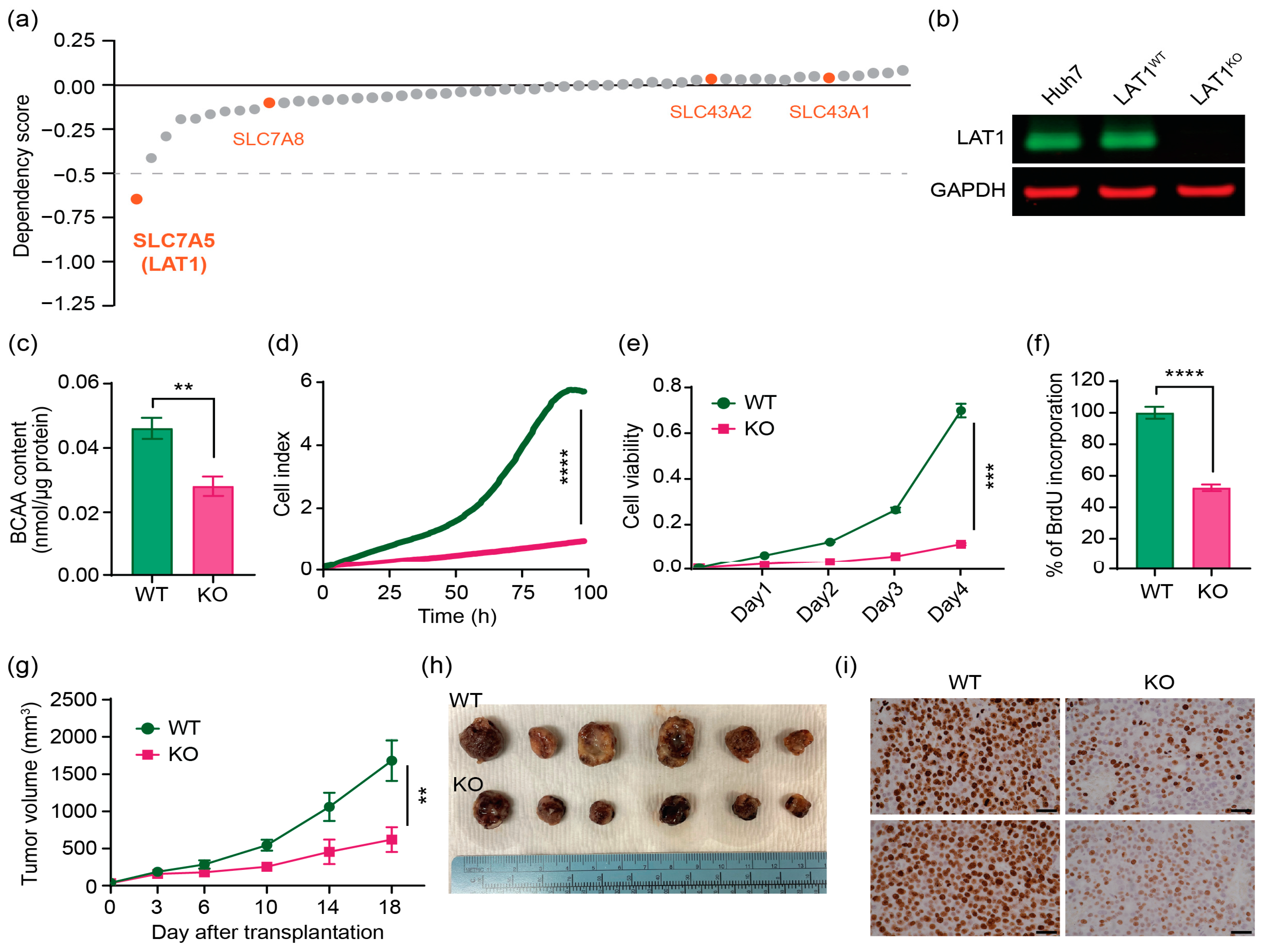
2.2. CRISPR/Cas9 Effectively Knocks out LAT1 Expression, Reduces BCAA Transport, and Suppresses Cell Proliferation In Vitro and in Mouse Xenograft Model
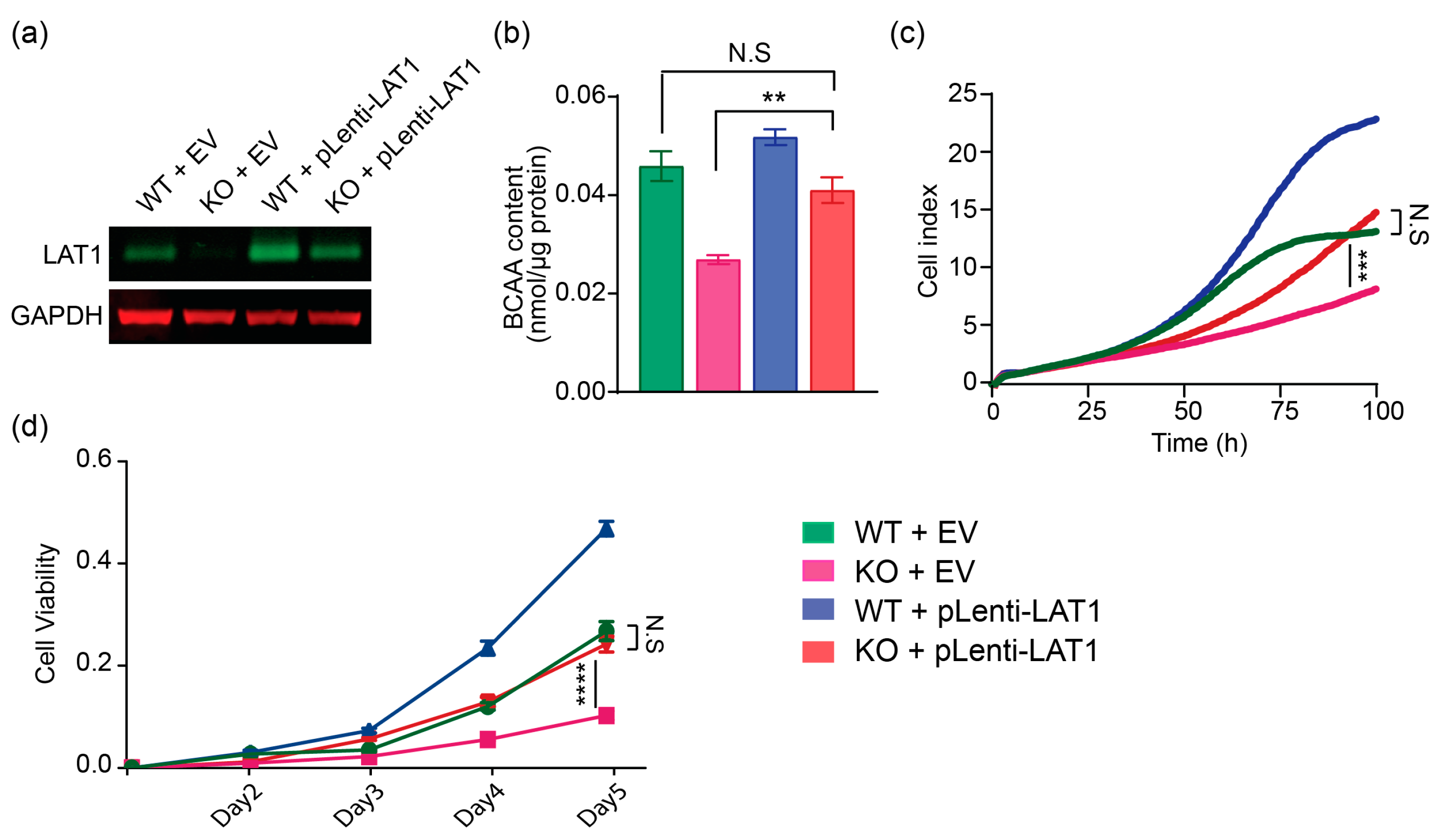
2.3. Lentiviral-LAT1 Infection Increases Cellular BCAA Levels and Rescues Cell Proliferation to Similar Levels as Cell Cultured in Wildtype Control Huh7 Cells
2.4. LAT1 Supports Proliferation of HCC by mTORC1 Pathway
3. Discussion
4. Materials and Methods
4.1. Guide RNA Design and Plasmid Construction
4.2. Cell Culture and sgRNA Transfection
4.3. CRISPR/Cas9 Mediated Gene Knockout of slc7a5
4.4. Cell Proliferation Assay
4.5. BCAA Content Measurement
4.6. Western Blot Analysis
4.7. Lentivirus Package and Transduction
4.8. Animal Studies and Animal Xenograft Models
4.9. Immunohostochemistry
4.10. Quantitative Real-Time PCR for Gene Expression Analysis
4.11. Short Hairprin Oligonucleotide Design
4.12. Public Data Availability and Processing
4.13. RNA Sequencing Analysis
4.14. Survival Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global Burden of Primary Liver Cancer in 2020 and Predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A Global View of Hepatocellular Carcinoma: Trends, Risk, Prevention and Management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Minaei, N.; Ramezankhani, R.; Tamimi, A.; Piryaei, A.; Zarrabi, A.; Aref, A.R.; Mostafavi, E.; Vosough, M. Immunotherapeutic Approaches in Hepatocellular Carcinoma: Building Blocks of Hope in near Future. Eur. J. Cell Biol. 2023, 102, 151284. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination Therapy with BPTES Nanoparticles and Metformin Targets the Metabolic Heterogeneity of Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and Cancer: Cell Biology, Physiology, and Clinical Opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; Van Den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the Therapeutic Response of Tumours to Dietary Serine and Glycine Starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Ding, Z.; Ericksen, R.E.; Escande-Beillard, N.; Lee, Q.Y.; Loh, A.; Denil, S.; Steckel, M.; Haegebarth, A.; Wai Ho, T.S.; Chow, P.; et al. Metabolic Pathway Analyses Identify Proline Biosynthesis Pathway as a Promoter of Liver Tumorigenesis. J. Hepatol. 2020, 72, 725–735. [Google Scholar] [CrossRef]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the MTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer Progression by Reprogrammed BCAA Metabolism in Myeloid Leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Wilkinson, A.C. Branched-Chain Amino Acid Metabolism in Cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Iwasa, M.; Tanaka, H.; Kaito, M.; Ikoma, J.; Shibata, T.; Takei, Y. Effect of Branched-Chain Amino Acids in Patients Receiving Intervention for Hepatocellular Carcinoma. World J. Gastroenterol. 2014, 20, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Shirakami, Y.; Hanai, T.; Imai, K.; Suetsugu, A.; Takai, K.; Shiraki, M.; Moriwaki, H. Pharmaceutical and Nutraceutical Approaches for Preventing Liver Carcinogenesis: Chemoprevention of Hepatocellular Carcinoma Using Acyclic Retinoid and Branched-Chain Amino Acids. Mol. Nutr. Food Res. 2014, 58, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, R.E.; Lim, S.L.; McDonnell, E.; Shuen, W.H.; Vadiveloo, M.; White, P.J.; Ding, Z.; Kwok, R.; Lee, P.; Radda, G.K.; et al. Loss of BCAA Catabolism during Carcinogenesis Enhances MTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab. 2019, 29, 1151–1165.e6. [Google Scholar] [CrossRef] [PubMed]
- Hyde, R.; Taylor, P.M.; Hundal, H.S. Amino Acid Transporters: Roles in Amino Acid Sensing and Signalling in Animal Cells. Biochem. J. 2003, 373, 1–18. [Google Scholar] [CrossRef]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.; Li, D.; Fu, L.; Zhang, X.; Bao, Y.; Zheng, L. Review of the Correlation of LAT1 With Diseases: Mechanism and Treatment. Front. Chem. 2020, 8, 967. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.; Pan, J. The Role of L-Type Amino Acid Transporter 1 in Human Tumors. Intractable Rare Dis. Res. 2015, 4, 165. [Google Scholar] [CrossRef]
- Häfliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1—An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutté, E.; Massard, P.A.; De La Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of MTORC1 and Tumor Growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bailey, C.G.; Ng, C.; Tiffen, J.; Thoeng, A.; Minhas, V.; Lehman, M.L.; Hendy, S.C.; Buchanan, G.; Nelson, C.C.; et al. Androgen Receptor and Nutrient Signaling Pathways Coordinate the Demand for Increased Amino Acid Transport during Prostate Cancer Progression. Cancer Res. 2011, 71, 7525–7536. [Google Scholar] [CrossRef] [PubMed]
- El Ansari, R.; Craze, M.L.; Miligy, I.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. The Amino Acid Transporter SLC7A5 Confers a Poor Prognosis in the Highly Proliferative Breast Cancer Subtypes and Is a Key Therapeutic Target in Luminal B Tumours. Breast Cancer Res. 2018, 20, 21. [Google Scholar] [CrossRef]
- Toyoshima, J.; Kusuhara, H.; Wempe, M.F.; Endou, H.; Sugiyama, Y. Investigation of the Role of Transporters on the Hepatic Elimination of an LAT1 Selective Inhibitor JPH203. J. Pharm. Sci. 2013, 102, 3228–3238. [Google Scholar] [CrossRef] [PubMed]
- Brunocilla, C.; Console, L.; Rovella, F.; Indiveri, C. Insights into the Transport Cycle of LAT1 and Interaction with the Inhibitor JPH203. Int. J. Mol. Sci. 2023, 24, 4042. [Google Scholar] [CrossRef]
- Okunushi, K.; Furihata, T.; Morio, H.; Muto, Y.; Higuchi, K.; Kaneko, M.; Otsuka, Y.; Ohno, Y.; Watanabe, Y.; Reien, Y.; et al. JPH203, a Newly Developed Anti-Cancer Drug, Shows a Preincubation Inhibitory Effect on L-Type Amino Acid Transporter 1 Function. J. Pharmacol. Sci. 2020, 144, 16–22. [Google Scholar] [CrossRef]
- Choi, D.W.; Kim, D.K.; Kanai, Y.; Wempe, M.F.; Endou, H.; Kim, J.K. JPH203, a Selective L-Type Amino Acid Transporter 1 Inhibitor, Induces Mitochondria-Dependent Apoptosis in Saos2 Human Osteosarcoma Cells. Korean J. Physiol. Pharmacol. 2017, 21, 599–607. [Google Scholar] [CrossRef]
- Wang, Q.; Tiffen, J.; Bailey, C.G.; Lehman, M.L.; Ritchie, W.; Fazli, L.; Metierre, C.; Feng, Y.J.; Li, E.; Gleave, M.; et al. Targeting Amino Acid Transport in Metastatic Castration-Resistant Prostate Cancer: Effects on Cell Cycle, Cell Growth, and Tumor Development. J. Natl. Cancer Inst. 2013, 105, 1463–1473. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 Is a Leucine Sensor for the MTORC1 Pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef]
- Dyachok, J.; Earnest, S.; Iturraran, E.N.; Cobb, M.H.; Ross, E.M. Amino Acids Regulate MTORC1 by an Obligate Two-Step Mechanism. J. Biol. Chem. 2016, 291, 22414–22426. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, P.J.; Kron, C.D.; Wittke, E.F.; Czerniak, B.N.; Bode, B.P. Targeted Suppression and Knockout of ASCT2 or LAT1 in Epithelial and Mesenchymal Human Liver Cancer Cells Fail to Inhibit Growth. Int. J. Mol. Sci. 2018, 19, 2093. [Google Scholar] [CrossRef]
- Cui, D.; Qu, R.; Liu, D.; Xiong, X.; Liang, T.; Zhao, Y. The Cross Talk Between P53 and MTOR Pathways in Response to Physiological and Genotoxic Stresses. Front. Cell Dev. Biol. 2021, 9, 3320. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- Wang, Q.; Holst, J. L-Type Amino Acid Transport and Cancer: Targeting the MTORC1 Pathway to Inhibit Neoplasia. Am. J. Cancer Res. 2015, 5, 1281. [Google Scholar] [PubMed]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Okuda, K.; Umemura, A.; Kataoka, S.; Yano, K.; Takahashi, A.; Okishio, S.; Taketani, H.; Seko, Y.; Nishikawa, T.; Yamaguchi, K.; et al. Enhanced Antitumor Effect in Liver Cancer by Amino Acid Depletion-Induced Oxidative Stress. Front. Oncol. 2021, 11, 758549. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, C.; Liu, Z. Essential Amino Acids Deprivation Is a Potential Strategy for Breast Cancer Treatment. Breast 2022, 62, 152–161. [Google Scholar] [CrossRef]
- Ali, E.S.; Mitra, K.; Akter, S.; Ramproshad, S.; Mondal, B.; Khan, I.N.; Islam, M.T.; Sharifi-Rad, J.; Calina, D.; Cho, W.C. Recent Advances and Limitations of MTOR Inhibitors in the Treatment of Cancer. Cancer Cell Int. 2022, 22, 284. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Paliogiannis, P.; Calvisi, D.F.; Chen, X. Role of the MTOR Pathway in Liver Cancer: From Molecular Genetics to Targeted Therapies. Hepatology 2021, 73, 49. [Google Scholar] [CrossRef]
- Gatto, L.K.T. depmap: Cancer Dependency Map Data Package. R Package Version 1.14.0. 2023. Available online: https://bioconductor.org/packages/release/data/experiment/manuals/depmap/man/depmap.pdf (accessed on 27 April 2023).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Ouyang, J.; Qin, G.; Liu, Z.; Jian, X.; Shi, T.; Xie, L. ToPP: Tumor Online Prognostic Analysis Platform for Prognostic Feature Selection and Clinical Patient Subgroup Selection. iScience 2022, 25, 104190. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-Y.; Ong, Q.; Liao, Y.; Ding, Z.; Tan, A.Q.L.; Lim, L.T.R.; Tan, H.M.; Lim, S.L.; Lee, Q.Y.; Han, W. Genetic Ablation of LAT1 Inhibits Growth of Liver Cancer Cells and Downregulates mTORC1 Signaling. Int. J. Mol. Sci. 2023, 24, 9171. https://doi.org/10.3390/ijms24119171
Kim S-Y, Ong Q, Liao Y, Ding Z, Tan AQL, Lim LTR, Tan HM, Lim SL, Lee QY, Han W. Genetic Ablation of LAT1 Inhibits Growth of Liver Cancer Cells and Downregulates mTORC1 Signaling. International Journal of Molecular Sciences. 2023; 24(11):9171. https://doi.org/10.3390/ijms24119171
Chicago/Turabian StyleKim, Sun-Yee, Qunxiang Ong, Yilie Liao, Zhaobing Ding, Alicia Qian Ler Tan, Ler Ting Rachel Lim, Hui Min Tan, Siew Lan Lim, Qian Yi Lee, and Weiping Han. 2023. "Genetic Ablation of LAT1 Inhibits Growth of Liver Cancer Cells and Downregulates mTORC1 Signaling" International Journal of Molecular Sciences 24, no. 11: 9171. https://doi.org/10.3390/ijms24119171
APA StyleKim, S.-Y., Ong, Q., Liao, Y., Ding, Z., Tan, A. Q. L., Lim, L. T. R., Tan, H. M., Lim, S. L., Lee, Q. Y., & Han, W. (2023). Genetic Ablation of LAT1 Inhibits Growth of Liver Cancer Cells and Downregulates mTORC1 Signaling. International Journal of Molecular Sciences, 24(11), 9171. https://doi.org/10.3390/ijms24119171