
Microglia and the Blood–Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
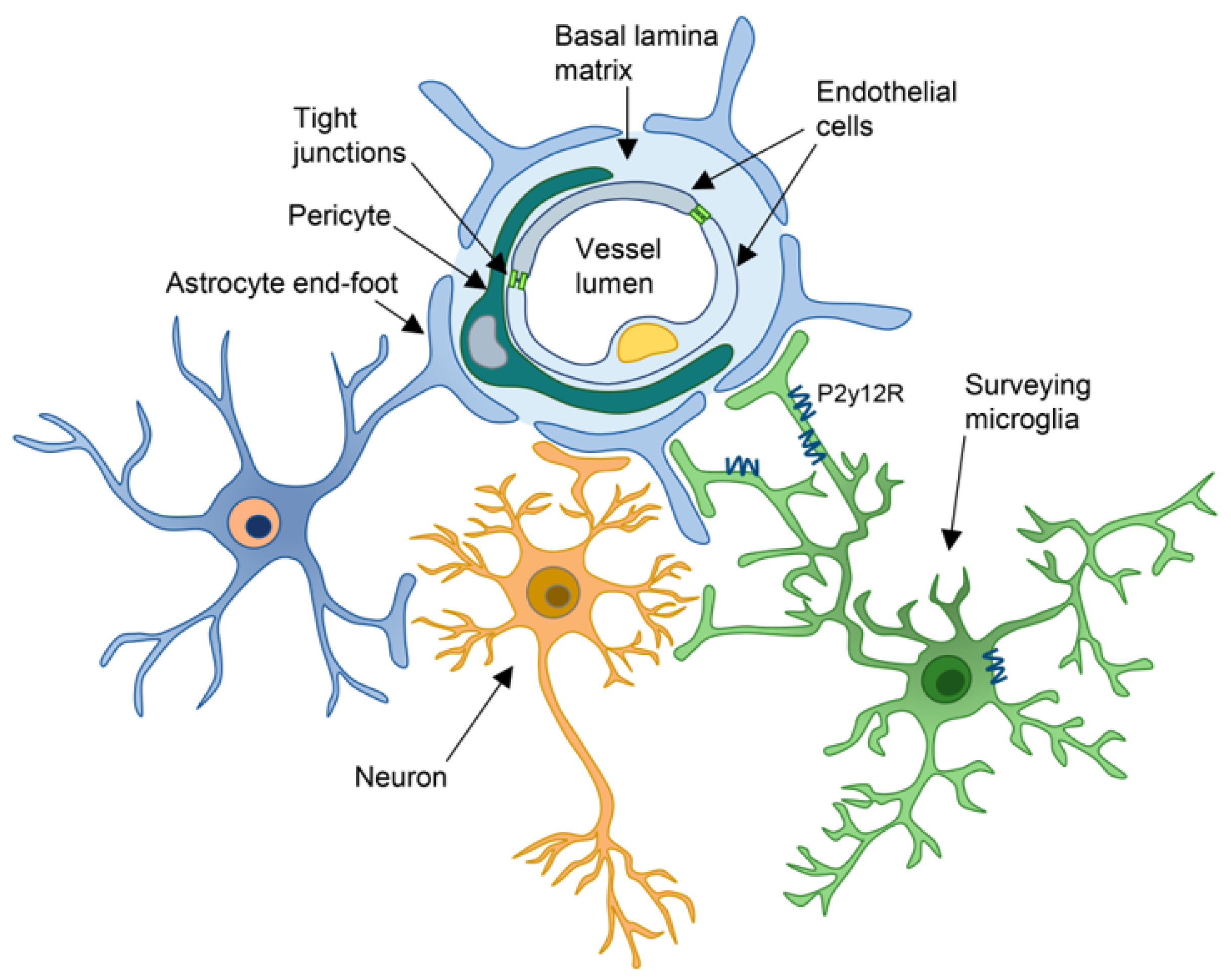
2. Components of the NVU
2.1. Endothelial Cells
2.2. Pericytes
2.3. Astrocytes
2.4. Microglia
3. Acute Injury: How Microglia Affect BBB Properties in Ischemic Stroke
3.1. Microglia–Neuron Crosstalk
3.2. Microglia–Endothelial Cell Crosstalk
3.3. Microglia–Astrocyte Crosstalk
3.4. Microglia–Peripheral Leukocyte Crosstalk
4. Microglia in a Chronic Inflammatory Condition: Alzheimer’s Disease
4.1. The Impairment of Endothelial Cells at the BBB in Alzheimer’s Disease
4.2. Microglia Role in AD
4.3. Astrocyte Role in AD and Their Crosstalk with Microglia
4.4. Pericyte Crosstalk with Microglia in AD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Brightman, M.W.; Reese, T.S. Junctions between intimately apposed cell membranes in the vertebrate brain. J. Cell Biol. 1969, 40, 648–677. [Google Scholar] [CrossRef]
- Westergaard, E.; Brightman, M.W. Transport of proteins across normal cerebral arterioles. J. Comp. Neurol. 1973, 152, 17–44. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Dore-Duffy, P. Role of the CNS microvascular pericyte in the blood-brain barrier. J. Neurosci. Res. 1998, 53, 637–644. [Google Scholar] [CrossRef]
- Del Zoppo, G.J.; Milner, R.; Mabuchi, T.; Hung, S.; Wang, X.; Koziol, J.A. Vascular matrix adhesion and the blood-brain barrier. Biochem. Soc. Trans. 2006, 34 Pt 6, 1261–1266. [Google Scholar] [CrossRef]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Patton, B.L. Developmental and pathogenic mechanisms of basement membrane assembly. Curr. Pharm. Des. 2009, 15, 1277–1294. [Google Scholar] [CrossRef]
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics 2008, 5, 409–414. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40 (Suppl. S1), S6–S24. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Yang, J.; Ronaldson, P.T.; Davis, T.P. Structure, Function, and Regulation of the Blood-Brain Barrier Tight Junction in Central Nervous System Disorders. Front. Physiol. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Yamaguchi, H.; Katsukura, Y.; Asashima, T.; Terasaki, T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J. Neurochem. 2008, 104, 147–154. [Google Scholar] [CrossRef]
- Buschmann, M.M.; Shen, L.; Rajapakse, H.; Raleigh, D.R.; Wang, Y.; Wang, Y.; Lingaraju, A.; Zha, J.; Abbott, E.; McAuley, E.M.; et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol. Biol. Cell 2013, 24, 3056–3068. [Google Scholar] [CrossRef]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef]
- Shin, K.; Margolis, B. ZOning out tight junctions. Cell 2006, 126, 647–649. [Google Scholar] [CrossRef]
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert. Opin. Drug Deliv. 2019, 16, 271–285. [Google Scholar] [CrossRef]
- Puris, E.; Fricker, G.; Gynther, M. Targeting Transporters for Drug Delivery to the Brain: Can We Do Better? Pharm. Res. 2022, 39, 1415–1455. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef] [PubMed]
- Bohannon, D.G.; Long, D.; Kim, W.K. Understanding the Heterogeneity of Human Pericyte Subsets in Blood-Brain Barrier Homeostasis and Neurological Diseases. Cells 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Gautam, J.; Cao, Y.; Yao, Y. Pericytic Laminin Maintains Blood-Brain Barrier Integrity in an Age-Dependent Manner. Transl. Stroke Res. 2020, 11, 228–242. [Google Scholar] [CrossRef]
- Uemura, M.T.; Maki, T.; Ihara, M.; Lee, V.M.Y.; Trojanowski, J.Q. Brain Microvascular Pericytes in Vascular Cognitive Impairment and Dementia. Front. Aging Neurosci. 2020, 12, 80. [Google Scholar] [CrossRef]
- Mae, M.A.; He, L.; Nordling, S.; Vazquez-Liebanas, E.; Nahar, K.; Jung, B.; Li, X.; Tan, B.C.; Chin Foo, J.; Cazenave-Gassiot, A.; et al. Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss. Circ. Res. 2021, 128, e46–e62. [Google Scholar] [CrossRef]
- Quaegebeur, A.; Segura, I.; Carmeliet, P. Pericytes: Blood-brain barrier safeguards against neurodegeneration? Neuron 2010, 68, 321–323. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Cai, C.; Fordsmann, J.C.; Jensen, S.H.; Gesslein, B.; Lonstrup, M.; Hald, B.O.; Zambach, S.A.; Brodin, B.; Lauritzen, M.J. Stimulation-induced increases in cerebral blood flow and local capillary vasoconstriction depend on conducted vascular responses. Proc. Natl. Acad. Sci. USA 2018, 115, E5796–E5804. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E. The role of pericytes in angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, M.; Gerhardt, H.; Kalen, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001, 153, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Geranmayeh, M.H.; Rahbarghazi, R.; Farhoudi, M. Targeting pericytes for neurovascular regeneration. Cell Commun. Signal. 2019, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, A.; Tanaka, K.; Niwa, M. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, Z.L.; Norris, E.H.; Strickland, S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun. 2014, 5, 3413. [Google Scholar] [CrossRef]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The blood-brain barrier. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 133, pp. 39–59. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Bortolotto, V.; Canonico, P.L.; Sortino, M.A.; Grilli, M. Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood-Brain Barrier Integrity. Front. Pharmacol. 2019, 10, 1346. [Google Scholar] [CrossRef]
- Lecuyer, M.A.; Kebir, H.; Prat, A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochim. Biophys. Acta 2016, 1862, 472–482. [Google Scholar] [CrossRef]
- Kubotera, H.; Ikeshima-Kataoka, H.; Hatashita, Y.; Allegra Mascaro, A.L.; Pavone, F.S.; Inoue, T. Astrocytic endfeet re-cover blood vessels after removal by laser ablation. Sci. Rep. 2019, 9, 1263. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef] [PubMed]
- Dowell, J.A.; Johnson, J.A.; Li, L. Identification of astrocyte secreted proteins with a combination of shotgun proteomics and bioinformatics. J. Proteome Res. 2009, 8, 4135–4143. [Google Scholar] [CrossRef]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter Release from Astrocytes: Functional, Developmental, and Pathological Implications in the Brain. Front. Neurosci. 2015, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chang, L.; Wei, C. The sonic hedgehog pathway mediates Tongxinluo capsule-induced protection against blood-brain barrier disruption after ischaemic stroke in mice. Basic Clin. Pharmacol. Toxicol. 2019, 124, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Mizee, M.R.; Wooldrik, D.; Lakeman, K.A.; van het Hof, B.; Drexhage, J.A.; Geerts, D.; Bugiani, M.; Aronica, E.; Mebius, R.E.; Prat, A.; et al. Retinoic acid induces blood-brain barrier development. J. Neurosci. 2013, 33, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, R.L.; Hawelu-Johnson, C.L.; Guyenet, P.G.; Lynch, K.R. Astrocytes synthesize angiotensinogen in brain. Science 1988, 242, 1444–1446. [Google Scholar] [CrossRef]
- Wosik, K.; Cayrol, R.; Dodelet-Devillers, A.; Berthelet, F.; Bernard, M.; Moumdjian, R.; Bouthillier, A.; Reudelhuber, T.L.; Prat, A. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: Relevance to multiple sclerosis. J. Neurosci. 2007, 27, 9032–9042. [Google Scholar] [CrossRef]
- Takane, K.; Hasegawa, Y.; Lin, B.; Koibuchi, N.; Cao, C.; Yokoo, T.; Kim-Mitsuyama, S. Detrimental Effects of Centrally Administered Angiotensin II are Enhanced in a Mouse Model of Alzheimer Disease Independently of Blood Pressure. J. Am. Heart Assoc. 2017, 6, e004897. [Google Scholar] [CrossRef]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111 Pt 13, 1853–1865. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Avila, M.; Gonzalez, J.; El-Bacha, R.S.; Baez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front. Cell Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [PubMed]
- Mills, W.A., III; Woo, A.M.; Jiang, S.; Martin, J.; Surendran, D.; Bergstresser, M.; Kimbrough, I.F.; Eyo, U.B.; Sofroniew, M.V.; Sontheimer, H. Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat. Commun. 2022, 13, 1794. [Google Scholar] [CrossRef]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Sierra, A.; de Castro, F.; Del Rio-Hortega, J.; Rafael Iglesias-Rozas, J.; Garrosa, M.; Kettenmann, H. The “Big-Bang” for modern glial biology: Translation and comments on Pio del Rio-Hortega 1919 series of papers on microglia. Glia 2016, 64, 1801–1840. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics During Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Hagemeyer, N.; Kierdorf, K.; Frenzel, K.; Xue, J.; Ringelhan, M.; Abdullah, Z.; Godin, I.; Wieghofer, P.; Costa Jordao, M.J.; Ulas, T.; et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J. 2016, 35, 1730–1744. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef]
- George, J.; Cunha, R.A.; Mulle, C.; Amedee, T. Microglia-derived purines modulate mossy fibre synaptic transmission and plasticity through P2X4 and A1 receptors. Eur. J. Neurosci. 2016, 43, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Morganti, J.M.; Jernberg, J.; Schlunk, A.; Mitchell, S.H.; Brewster, K.W.; Hudson, C.E.; Cole, M.J.; Harrison, J.K.; Bickford, P.C.; et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 2011, 32, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.K.; Lifshitz, J. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Q.; Zhu, H.; Hou, L.; Liu, W.; Yang, Q.; Shen, H.; Chai, G.; Zhang, B.; Chen, S.; et al. Microglial Piezo1 senses Abeta fibril stiffness to restrict Alzheimer’s disease. Neuron 2023, 111, 15–29 e18. [Google Scholar] [CrossRef]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef]
- Cockram, T.O.J.; Puigdellivol, M.; Brown, G.C. Calreticulin and Galectin-3 Opsonise Bacteria for Phagocytosis by Microglia. Front. Immunol. 2019, 10, 2647. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leak, R.K.; Cao, G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front. Cell. Neurosci. 2022, 16, 980722. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Penna, E.; Mangum, J.M.; Shepherd, H.; Martinez-Cerdeno, V.; Noctor, S.C. Development of the Neuro-Immune-Vascular Plexus in the Ventricular Zone of the Prenatal Rat Neocortex. Cereb. Cortex 2021, 31, 2139–2155. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.A.; Greferath, U.; Fletcher, E.L.; Jobling, A.I. The Contribution of Microglia to the Development and Maturation of the Visual System. Front. Cell. Neurosci. 2021, 15, 659843. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 2009, 206, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Okojie, K.A.; Sharma, K.; Lentferink, D.H.; Sun, Y.Y.; Chen, H.R.; Uweru, J.O.; Amancherla, S.; Calcuttawala, Z.; Campos-Salazar, A.B.; et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat. Commun. 2021, 12, 5289. [Google Scholar] [CrossRef]
- Grossmann, R.; Stence, N.; Carr, J.; Fuller, L.; Waite, M.; Dailey, M.E. Juxtavascular microglia migrate along brain microvessels following activation during early postnatal development. Glia 2002, 37, 229–240. [Google Scholar] [CrossRef]
- Mondo, E.; Becker, S.C.; Kautzman, A.G.; Schifferer, M.; Baer, C.E.; Chen, J.; Huang, E.J.; Simons, M.; Schafer, D.P. A Developmental Analysis of Juxtavascular Microglia Dynamics and Interactions with the Vasculature. J. Neurosci. 2020, 40, 6503–6521. [Google Scholar] [CrossRef]
- Csaszar, E.; Lenart, N.; Cserep, C.; Kornyei, Z.; Fekete, R.; Posfai, B.; Balazsfi, D.; Hangya, B.; Schwarcz, A.D.; Szabadits, E.; et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J. Exp. Med. 2022, 219, e20211071. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, A.R.; Korolainen, M.A.; Odero, G.; Miller, D.W.; Kauppinen, T.M. Poly(ADP-ribose) polymerase-1 regulates microglia mediated decrease of endothelial tight junction integrity. Neurochem. Int. 2017, 108, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Guada, L.; Yavagal, D.R. Global Epidemiology of Stroke and Access to Acute Ischemic Stroke Interventions. Neurology 2021, 97 (Suppl. S2), S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Hata, R.; Maeda, K.; Hermann, D.; Mies, G.; Hossmann, K.A. Evolution of brain infarction after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2000, 20, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Castro, S.; Sousa, J.A.; Bras, A.; Cecilia, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood-Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, J.; Chang, J.Y.; Kim, S.H.; Lee, J.E. Inflammation after Ischemic Stroke: The Role of Leukocytes and Glial Cells. Exp. Neurobiol. 2016, 25, 241–251. [Google Scholar] [CrossRef]
- Hossmann, K.A. Pathophysiology and therapy of experimental stroke. Cell. Mol. Neurobiol. 2006, 26, 1057–1083. [Google Scholar] [CrossRef]
- Wieloch, T.; Nikolich, K. Mechanisms of neural plasticity following brain injury. Curr. Opin. Neurobiol. 2006, 16, 258–264. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef]
- Panayi, G.S.; Corrigall, V.M.; Henderson, B. Stress cytokines: Pivotal proteins in immune regulatory networks: Opinion. Curr. Opin. Immunol. 2004, 16, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.; Le Feuvre, R.A.; Iwakura, Y.; Rothwell, N.J. Purinergic (P2X7) receptor activation of microglia induces cell death via an interleukin-1-independent mechanism. Mol. Cell. Neurosci. 2002, 19, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Ransohoff, R.M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Front. Cell. Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kohsaka, S. Microglia: Neuroprotective and neurotrophic cells in the central nervous system. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2004, 4, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Surugiu, R.; Catalin, B.; Dumbrava, D.; Gresita, A.; Olaru, D.G.; Hermann, D.M.; Popa-Wagner, A. Intracortical Administration of the Complement C3 Receptor Antagonist Trifluoroacetate Modulates Microglia Reaction after Brain Injury. Neural Plast. 2019, 2019, 1071036. [Google Scholar] [CrossRef]
- Meng, H.L.; Li, X.X.; Chen, Y.T.; Yu, L.J.; Zhang, H.; Lao, J.M.; Zhang, X.; Xu, Y. Neuronal Soluble Fas Ligand Drives M1-Microglia Polarization after Cerebral Ischemia. CNS Neurosci. Ther. 2016, 22, 771–781. [Google Scholar] [CrossRef]
- Yin, Z.; Han, Z.; Hu, T.; Zhang, S.; Ge, X.; Huang, S.; Wang, L.; Yu, J.; Li, W.; Wang, Y.; et al. Neuron-derived exosomes with high miR-21-5p expression promoted polarization of M1 microglia in culture. Brain Behav. Immun. 2020, 83, 270–282. [Google Scholar] [CrossRef]
- Khatri, R.; McKinney, A.M.; Swenson, B.; Janardhan, V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology 2012, 79 (Suppl. S1), S52–S57. [Google Scholar] [CrossRef]
- Sarvari, S.; Moakedi, F.; Hone, E.; Simpkins, J.W.; Ren, X. Mechanisms in blood-brain barrier opening and metabolism-challenged cerebrovascular ischemia with emphasis on ischemic stroke. Metab. Brain Dis. 2020, 35, 851–868. [Google Scholar] [CrossRef]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, I.; Cuartero, M.I.; Pradillo, J.M.; Moro, M.A.; Lizasoain, I. Cytokines and Chemokines in stroke. In Cerebrovascular Diseases, 2nd ed.; Elsevier: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef]
- Xie, L.; Zhao, H.; Wang, Y.; Chen, Z. Exosomal shuttled miR-424-5p from ischemic preconditioned microglia mediates cerebral endothelial cell injury through negatively regulation of FGF2/STAT3 pathway. Exp. Neurol. 2020, 333, 113411. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, J.F.; Sarkar, S.; Tranter, K.M.; Hanig, J.P.; Miller, D.B.; O’Callaghan, J.P. Vascular-directed responses of microglia produced by methamphetamine exposure: Indirect evidence that microglia are involved in vascular repair? J. Neuroinflamm. 2016, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, J.; Ilkowski, J.; Losy, J. Serial measurements of levels of the chemokines CCL2, CCL3 and CCL5 in serum of patients with acute ischaemic stroke. Folia Neuropathol. 2006, 44, 282–289. [Google Scholar]
- Lee, J.Y.; Choi, H.Y.; Yune, T.Y. MMP-3 secreted from endothelial cells of blood vessels after spinal cord injury activates microglia, leading to oligodendrocyte cell death. Neurobiol. Dis. 2015, 82, 141–151. [Google Scholar] [CrossRef]
- Xu, Z.; Han, K.; Chen, J.; Wang, C.; Dong, Y.; Yu, M.; Bai, R.; Huang, C.; Hou, L. Vascular endothelial growth factor is neuroprotective against ischemic brain injury by inhibiting scavenger receptor A expression on microglia. J. Neurochem. 2017, 142, 700–709. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Austin, S.A. Neurovascular Protective Function of Endothelial Nitric Oxide—Recent Advances. Circ. J. 2016, 80, 1499–1503. [Google Scholar] [CrossRef]
- Pankratova, S.; Bjornsdottir, H.; Christensen, C.; Zhang, L.; Li, S.; Dmytriyeva, O.; Bock, E.; Berezin, V. Immunomodulator CD200 Promotes Neurotrophic Activity by Interacting with and Activating the Fibroblast Growth Factor Receptor. Mol. Neurobiol. 2016, 53, 584–594. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, C.; Arrick, D.M.; Yang, S.; Baluna, A.E.; Sun, H. Role of nitric oxide synthases in early blood-brain barrier disruption following transient focal cerebral ischemia. PLoS ONE 2014, 9, e93134. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2019, 39, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Chen, A.Y.; Hung, V.K.; Yaw, L.P.; Fung, M.K.; Ho, M.C.; Tsang, M.C.; Chung, S.S.; Chung, S.K. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. J. Cereb. Blood Flow Metab. 2005, 25, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1beta induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef]
- Zhang, Y.; Lian, L.; Fu, R.; Liu, J.; Shan, X.; Jin, Y.; Xu, S. Microglia: The Hub of Intercellular Communication in Ischemic Stroke. Front. Cell. Neurosci. 2022, 16, 889442. [Google Scholar] [CrossRef]
- He, M.; Dong, H.; Huang, Y.; Lu, S.; Zhang, S.; Qian, Y.; Jin, W. Astrocyte-Derived CCL2 is Associated with M1 Activation and Recruitment of Cultured Microglial Cells. Cell. Physiol. Biochem. 2016, 38, 859–870. [Google Scholar] [CrossRef]
- Xue, J.; Zhang, Y.; Zhang, J.; Zhu, Z.; Lv, Q.; Su, J. Astrocyte-derived CCL7 promotes microglia-mediated inflammation following traumatic brain injury. Int. Immunopharmacol. 2021, 99, 107975. [Google Scholar] [CrossRef]
- Kim, S.; Son, Y. Astrocytes Stimulate Microglial Proliferation and M2 Polarization In Vitro through Crosstalk between Astrocytes and Microglia. Int. J. Mol. Sci. 2021, 22, 8800. [Google Scholar] [CrossRef]
- Min, K.J.; Yang, M.S.; Kim, S.U.; Jou, I.; Joe, E.H. Astrocytes induce hemeoxygenase-1 expression in microglia: A feasible mechanism for preventing excessive brain inflammation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep. 2021, 34, 108882. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Fenn, A.M.; Dugan, A.; Godbout, J.P. TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 2014, 62, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Planas, A.M. Role of Immune Cells Migrating to the Ischemic Brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Gullotta, G.S.; De Feo, D.; Friebel, E.; Semerano, A.; Scotti, G.M.; Bergamaschi, A.; Butti, E.; Brambilla, E.; Genchi, A.; Capotondo, A.; et al. Age-induced alterations of granulopoiesis generate atypical neutrophils that aggravate stroke pathology. Nat. Immunol. 2023. [Google Scholar] [CrossRef]
- Neumann, J.; Sauerzweig, S.; Ronicke, R.; Gunzer, F.; Dinkel, K.; Ullrich, O.; Gunzer, M.; Reymann, K.G. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: A new mechanism of CNS immune privilege. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 5965–5975. [Google Scholar] [CrossRef]
- Otxoa-de-Amezaga, A.; Miro-Mur, F.; Pedragosa, J.; Gallizioli, M.; Justicia, C.; Gaja-Capdevila, N.; Ruiz-Jaen, F.; Salas-Perdomo, A.; Bosch, A.; Calvo, M.; et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019, 137, 321–341. [Google Scholar] [CrossRef]
- Kim, Y.R.; Kim, Y.M.; Lee, J.; Park, J.; Lee, J.E.; Hyun, Y.M. Neutrophils Return to Bloodstream Through the Brain Blood Vessel After Crosstalk With Microglia during LPS-Induced Neuroinflammation. Front. Cell Dev. Biol. 2020, 8, 613733. [Google Scholar] [CrossRef]
- Gan, Y.; Liu, Q.; Wu, W.; Yin, J.X.; Bai, X.F.; Shen, R.; Wang, Y.; Chen, J.; La Cava, A.; Poursine-Laurent, J.; et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc. Natl. Acad. Sci. USA 2014, 111, 2704–2709. [Google Scholar] [CrossRef]
- Lunemann, A.; Lunemann, J.D.; Roberts, S.; Messmer, B.; Barreira da Silva, R.; Raine, C.S.; Munz, C. Human NK cells kill resting but not activated microglia via NKG2D- and NKp46-mediated recognition. J. Immunol. 2008, 181, 6170–6177. [Google Scholar] [CrossRef]
- Lee, G.A.; Lin, T.N.; Chen, C.Y.; Mau, S.Y.; Huang, W.Z.; Kao, Y.C.; Ma, R.Y.; Liao, N.S. Interleukin 15 blockade protects the brain from cerebral ischemia-reperfusion injury. Brain Behav. Immun. 2018, 73, 562–570. [Google Scholar] [CrossRef]
- Korhonen, P.; Kanninen, K.M.; Lehtonen, S.; Lemarchant, S.; Puttonen, K.A.; Oksanen, M.; Dhungana, H.; Loppi, S.; Pollari, E.; Wojciechowski, S.; et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav. Immun. 2015, 49, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Chabot, S.; Charlet, D.; Wilson, T.L.; Yong, V.W. Cytokine production consequent to T cell—microglia interaction: The PMA/IFN gamma-treated U937 cells display similarities to human microglia. J. Neurosci. Methods 2001, 105, 111–120. [Google Scholar] [CrossRef]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhong, D.; Zheng, Y.; Li, H.; Chen, H.; Ma, S.; Sun, Y.; Yan, W.; Li, G. Damage effect of interleukin (IL)-23 on oxygen-glucose-deprived cells of the neurovascular unit via IL-23 receptor. Neuroscience 2015, 289, 406–416. [Google Scholar] [CrossRef]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- DiChiara, T.; DiNunno, N.; Clark, J.; Bu, R.L.; Cline, E.N.; Rollins, M.G.; Gong, Y.; Brody, D.L.; Sligar, S.G.; Velasco, P.T.; et al. Alzheimer’s Toxic Amyloid Beta Oligomers: Unwelcome Visitors to the Na/K ATPase alpha3 Docking Station. Yale J. Biol. Med. 2017, 90, 45–61. [Google Scholar]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef]
- Texido, L.; Martin-Satue, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Merlo, S.; Spampinato, S.F.; Caruso, G.I.; Sortino, M.A. The Ambiguous Role of Microglia in Abeta Toxicity: Chances for Therapeutic Intervention. Curr. Neuropharmacol. 2020, 18, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Caruso, G.; Spampinato, S.F.; Cardaci, V.; Caraci, F.; Sortino, M.A.; Merlo, S. beta-amyloid and Oxidative Stress: Perspectives in Drug Development. Curr. Pharm. Des. 2019, 25, 4771–4781. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorle, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid beta Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.C.; Hsieh, Y.C.; Hu, C.J.; Tu, Y.K. Endothelial Dysfunction in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 2909. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Oikari, L.E.; Pandit, R.; Stewart, R.; Cuni-Lopez, C.; Quek, H.; Sutharsan, R.; Rantanen, L.M.; Oksanen, M.; Lehtonen, S.; de Boer, C.M.; et al. Altered Brain Endothelial Cell Phenotype from a Familial Alzheimer Mutation and Its Potential Implications for Amyloid Clearance and Drug Delivery. Stem Cell Rep. 2020, 14, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Kirabali, T.; Rust, R.; Rigotti, S.; Siccoli, A.; Nitsch, R.M.; Kulic, L. Distinct changes in all major components of the neurovascular unit across different neuropathological stages of Alzheimer’s disease. Brain Pathol. 2020, 30, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: The role, regulation and restoration of LRP1. Front. Aging Neurosci. 2015, 7, 136. [Google Scholar] [CrossRef]
- Alemi, M.; Gaiteiro, C.; Ribeiro, C.A.; Santos, L.M.; Gomes, J.R.; Oliveira, S.M.; Couraud, P.O.; Weksler, B.; Romero, I.; Saraiva, M.J.; et al. Transthyretin participates in beta-amyloid transport from the brain to the liver--involvement of the low-density lipoprotein receptor-related protein 1? Sci. Rep. 2016, 6, 20164. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Deane, R.; Zlokovic, B.V. Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2007, 4, 191–197. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007, 1181, 93–103. [Google Scholar] [CrossRef]
- Merlini, M.; Meyer, E.P.; Ulmann-Schuler, A.; Nitsch, R.M. Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 2011, 122, 293–311. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Huang, W.; Xia, Q.; Zheng, F.; Zhao, X.; Ge, F.; Xiao, J.; Liu, Z.; Shen, Y.; Ye, K.; Wang, D.; et al. Microglia-Mediated Neurovascular Unit Dysfunction in Alzheimer’s Disease. J. Alzheimers Dis. J. ahead of print. 2023. [Google Scholar]
- Kook, S.Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca2+-calcineurin signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Aβ(1–42) oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J. Neurochem. 2015, 134, 382–393. [Google Scholar] [CrossRef]
- Marco, S.; Skaper, S.D. Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci. Lett. 2006, 401, 219–224. [Google Scholar] [CrossRef]
- Gonzalez-Velasquez, F.J.; Kotarek, J.A.; Moss, M.A. Soluble aggregates of the amyloid-beta protein selectively stimulate permeability in human brain microvascular endothelial monolayers. J. Neurochem. 2008, 107, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Usatyuk, P.V.; Enika, D.; Natarajan, V.; Rifkind, J.M. Vascular endothelial barrier dysfunction mediated by amyloid-beta proteins. J. Alzheimers Dis. 2009, 17, 845–854. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Walsh, D.M.; O’Malley, T.; Hudson, N.; Crosbie, D.E.; Loftus, T.; Sheehan, F.; McDaid, J.; Humphries, M.M.; Callanan, J.J.; et al. Autoregulated paracellular clearance of amyloid-beta across the blood-brain barrier. Sci. Adv. 2015, 1, e1500472. [Google Scholar] [CrossRef]
- Schlingmann, B.; Overgaard, C.E.; Molina, S.A.; Lynn, K.S.; Mitchell, L.A.; Dorsainvil White, S.; Mattheyses, A.L.; Guidot, D.M.; Capaldo, C.T.; Koval, M. Regulation of claudin/zonula occludens-1 complexes by hetero-claudin interactions. Nat. Commun. 2016, 7, 12276. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D.J.; et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 2018, 141, 2740–2754. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Grutzendler, J. Microglia-Mediated Neuroprotection, TREM2, and Alzheimer’s Disease: Evidence From Optical Imaging. Biol. Psychiatry 2018, 83, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Czirr, E.; Castello, N.A.; Mosher, K.I.; Castellano, J.M.; Hinkson, I.V.; Lucin, K.M.; Baeza-Raja, B.; Ryu, J.K.; Li, L.; Farina, S.N.; et al. Microglial complement receptor 3 regulates brain Abeta levels through secreted proteolytic activity. J. Exp. Med. 2017, 214, 1081–1092. [Google Scholar] [CrossRef]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef] [PubMed]
- Merlo, S.; Spampinato, S.F.; Sortino, M.A. Early compensatory responses against neuronal injury: A new therapeutic window of opportunity for Alzheimer’s Disease? CNS Neurosci. Ther. 2019, 25, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.; Glabe, C.; Rogers, J. Multivalent binding of complement protein C1Q to the amyloid beta-peptide (A beta) promotes the nucleation phase of A beta aggregation. Biochem. Biophys. Res. Commun. 1995, 217, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Huffels, C.F.M.; Middeldorp, J.; Hol, E.M. Ass Pathology and Neuron-Glia Interactions: A Synaptocentric View. Neurochem. Res. 2023, 48, 1026–1046. [Google Scholar] [CrossRef] [PubMed]
- Sajja, V.S.; Hlavac, N.; VandeVord, P.J. Role of Glia in Memory Deficits Following Traumatic Brain Injury: Biomarkers of Glia Dysfunction. Front. Integr. Neurosci. 2016, 10, 7. [Google Scholar] [CrossRef]
- Okun, E.; Griffioen, K.J.; Lathia, J.D.; Tang, S.C.; Mattson, M.P.; Arumugam, T.V. Toll-like receptors in neurodegeneration. Brain Res. Rev. 2009, 59, 278–292. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Bolos, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lapointe, B.M.; Clark, S.R.; Zbytnuik, L.; Kubes, P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J. Immunol. 2006, 177, 8103–8110. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- McKim, D.B.; Weber, M.D.; Niraula, A.; Sawicki, C.M.; Liu, X.; Jarrett, B.L.; Ramirez-Chan, K.; Wang, Y.; Roeth, R.M.; Sucaldito, A.D.; et al. Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol. Psychiatry 2018, 23, 1421–1431. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Costantino, G.; Merlo, S.; Canonico, P.L.; Sortino, M.A. Microglia Contributes to BAF-312 Effects on Blood-Brain Barrier Stability. Biomolecules 2022, 12, 1174. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Kenkhuis, B.; Somarakis, A.; Kleindouwel, L.R.T.; van Roon-Mom, W.M.C.; Hollt, T.; van der Weerd, L. Co-expression patterns of microglia markers Iba1, TMEM119 and P2RY12 in Alzheimer’s disease. Neurobiol. Dis. 2022, 167, 105684. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Propson, N.E.; Gedam, M.; Zheng, H. Complement in Neurologic Disease. Annu. Rev. Pathol. 2021, 16, 277–298. [Google Scholar] [CrossRef]
- Domercq, M.; Brambilla, L.; Pilati, E.; Marchaland, J.; Volterra, A.; Bezzi, P. P2Y1 receptor-evoked glutamate exocytosis from astrocytes: Control by tumor necrosis factor-alpha and prostaglandins. J. Biol. Chem. 2006, 281, 30684–30696. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Crugnola, A.; Giaccone, G.; Capobianco, R.; Mangieri, M.; Kingston, A.E.; Bloc, A.; Bezzi, P.; et al. Defective tumor necrosis factor-alpha-dependent control of astrocyte glutamate release in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2005, 280, 42088–42096. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.S.; Ding, H.G.; Chen, S.L.; Liu, X.Q.; Deng, Y.Y.; Jiang, W.Q.; Li, Y.; Huang, L.Q.; Han, Y.L.; Wen, M.Y.; et al. Hypertonic saline mediates the NLRP3/IL-1beta signaling axis in microglia to alleviate ischemic blood-brain barrier permeability by downregulating astrocyte-derived VEGF in rats. CNS Neurosci. Ther. 2020, 26, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Meme, W.; Calvo, C.F.; Froger, N.; Ezan, P.; Amigou, E.; Koulakoff, A.; Giaume, C. Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: Potentiation by beta-amyloid. FASEB J. 2006, 20, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, W.; Middeldorp, J.; Kooijman, L.; Sluijs, J.A.; Kooi, E.J.; Moeton, M.; Freriks, M.; Mizee, M.R.; Hol, E.M. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 492–510. [Google Scholar] [CrossRef]
- Colombo, J.A.; Quinn, B.; Puissant, V. Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain Res. Bull. 2002, 58, 235–242. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef]
- Saavedra, J.; Nascimento, M.; Liz, M.A.; Cardoso, I. Key brain cell interactions and contributions to the pathogenesis of Alzheimer’s disease. Front. Cell Dev. Biol. 2022, 10, 1036123. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Abeta Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e1147. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired Autophagy in APOE4 Astrocytes. J. Alzheimers Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Merlo, S.; Sano, Y.; Kanda, T.; Sortino, M.A. Astrocytes contribute to Abeta-induced blood-brain barrier damage through activation of endothelial MMP9. J. Neurochem. 2017, 142, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nunez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef]
- Lau, S.F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef]
- Thuringer, D.; Garrido, C. Molecular chaperones in the brain endothelial barrier: Neurotoxicity or neuroprotection? FASEB J. 2019, 33, 11629–11639. [Google Scholar] [CrossRef]
- Pan, J.; Ma, N.; Zhong, J.; Yu, B.; Wan, J.; Zhang, W. Age-associated changes in microglia and astrocytes ameliorate blood-brain barrier dysfunction. Mol. Ther. Nucleic Acids 2021, 26, 970–986. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Merlo, S.; Fagone, E.; Fruciano, M.; Sano, Y.; Kanda, T.; Sortino, M.A. Reciprocal Interplay Between Astrocytes and CD4+ Cells Affects Blood-Brain Barrier and Neuronal Function in Response to beta Amyloid. Front. Mol. Neurosci. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Pietronigro, E.; Zenaro, E.; Bianca, V.D.; Dusi, S.; Terrabuio, E.; Iannoto, G.; Slanzi, A.; Ghasemi, S.; Nagarajan, R.; Piacentino, G.; et al. Blockade of alpha4 integrins reduces leukocyte-endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 12055. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Alcendor, D.J. Interactions between Amyloid-Beta Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1490. [Google Scholar] [CrossRef]
- Kitaguchi, H.; Ihara, M.; Saiki, H.; Takahashi, R.; Tomimoto, H. Capillary beds are decreased in Alzheimer’s disease, but not in Binswanger’s disease. Neurosci. Lett. 2007, 417, 128–131. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, 6450. [Google Scholar] [CrossRef]
- Takata, F.; Dohgu, S.; Matsumoto, J.; Takahashi, H.; Machida, T.; Wakigawa, T.; Harada, E.; Miyaji, H.; Koga, M.; Nishioku, T.; et al. Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-alpha, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflamm. 2011, 8, 106. [Google Scholar] [CrossRef]
- Savitz, S.I.; Cox, C.S., Jr. Concise Review: Cell Therapies for Stroke and Traumatic Brain Injury: Targeting Microglia. Stem Cells 2016, 34, 537–542. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gullotta, G.S.; Costantino, G.; Sortino, M.A.; Spampinato, S.F. Microglia and the Blood–Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions. Int. J. Mol. Sci. 2023, 24, 9144. https://doi.org/10.3390/ijms24119144
Gullotta GS, Costantino G, Sortino MA, Spampinato SF. Microglia and the Blood–Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions. International Journal of Molecular Sciences. 2023; 24(11):9144. https://doi.org/10.3390/ijms24119144
Chicago/Turabian StyleGullotta, Giorgia Serena, Giuseppe Costantino, Maria Angela Sortino, and Simona Federica Spampinato. 2023. "Microglia and the Blood–Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions" International Journal of Molecular Sciences 24, no. 11: 9144. https://doi.org/10.3390/ijms24119144
APA StyleGullotta, G. S., Costantino, G., Sortino, M. A., & Spampinato, S. F. (2023). Microglia and the Blood–Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions. International Journal of Molecular Sciences, 24(11), 9144. https://doi.org/10.3390/ijms24119144