1. Introduction
Chemotherapy is one of the most important pillars in the treatment of cancer diseases. However, the toxicity of this approach, resulting from the unspecific interaction of cytotoxic drugs with healthy tissue, presents one of the major drawbacks of this cancer treatment [
1,
2].
Targeted drug delivery is one of the strategies developed in recent decades to face this challenge. Antibody–Drug Conjugates (ADCs) represent a breakthrough [
3,
4]. An ADC consists of a cytotoxic drug and an antibody with high affinity to its oncological target. Both components are connected via a linker unit, which should be easily cleavable. Since ADCs are supposed to be stable constructs, the cytotoxic payload is only released after binding to the target. Nevertheless, this approach seems to have some limitations and disadvantages, such as a long circulation time and, therefore, high exposure for healthy tissues, reduced EPR effect, due to low penetration of the large antibodies into tumor tissue, and expensive and sophisticated synthesis of the drug conjugates [
4,
5,
6,
7]. Small molecule–drug conjugates (SMDCs) have been developed to tackle some of these challenges. Their low molecular weight enables good cell penetration in solid tumors. Moreover, as most SMDCs are smaller than 40 kDa, they are rapidly excreted from the blood through glomerular filtration, and, thus, display lower off-target cytotoxicity than ADCs. Other noteworthy facts concerning SMDCs are the simplified and manageable synthesis, compared to the sophisticated manufacturing process of monoclonal antibodies (mAbs), as well as the more straightforward transportation and storage processes, which are of immense value in regard to translational use of the corresponding drug conjugates [
8,
9].
Prostate cancer is the most common cancer in men and the second-leading cause of cancer death worldwide [
10,
11]. In the management of prostate cancer, the prostate-specific membrane antigen (PSMA) has been validated as a reliable tumor-associated biomarker and target for diagnosis, as well as for therapy of this disease [
12,
13]. PSMA is a membrane glycoprotein expressed on prostate tumor cells at significantly higher concentrations than on healthy tissue. Additionally, PSMA expression correlates with the grade of metastasis and progression of the tumor, allowing concise disease staging and therapy [
14,
15,
16].
In the last decade, several PSMA-targeting pharmaceuticals have been developed. Small molecule, radio-labeled PSMA inhibitor-based radiopharmaceuticals, such as [
68Ga]Ga-PSMA-11 and [
177Lu]Lu-PSMA-617, have played significant roles in the tremendous progress nuclear medicine has experienced in the diagnosis and therapy of prostate cancer [
17,
18,
19,
20]. However, only a few PSMA-targeted drug conjugates have been investigated so far.
Targeting PSMA via mAb was the strategy adopted in the development of MLN2704, which was the first clinically tested PSMA-ADC. MLN2704 consists of the mAb J591 conjugated to the microtubule inhibitor maytansinoid 1 via a redox-sensitive disulfide linker. Although MLN2704 displayed satisfying results in a phase 1 clinical trial conducted with 23 patients, further application was discontinued after conducting a phase 2 large cohort study, due to severe adverse effects, such as neurotoxicity. It was assumed that the instability of the ADC and the resulting premature release of the cytotoxic drug were the major reasons for this negative outcome [
21]. PSMA–MMAE is another PSMA-targeting ADC recently investigated in several clinical trials. The main component of PSMA–MMAE is the fully human mAb IgG1, which is conjugated to the antimitotic drug MMAE through a protease cleavable linker [
22]. Although the results of its phase 2 clinical trial, reported in 2020, indicated lower neurotoxicity than MLN2704, instability of the drug conjugates, and subsequent deconjugation, were the major limitations of further clinical application, even though an enzyme cleavable linker was used [
23,
24]. A similar fate was faced by MEDI3726, a PSMA–ADC with high specific cytotoxicity in vitro but insufficient tolerability in patients. Further development was discontinued after the outcomes of the phase 1 study [
25]. Generally, it can be concluded that the investigated PSMA–ADCs have failed, so far, in delivering the cytotoxic payload specifically to the tumor, due to their instability in blood circulation and the subsequent premature drug release. One possible approach to avoid these pitfalls is the development of small molecule–drug conjugates through replacement of the mAb in ADCs with small molecule inhibitors. Inspired by the successful implementation of this approach in PSMA radiopharmaceuticals, several efforts have been, and continue to be, made to apply the lessons learned from PSMA research.
Roy et al. [
26] described a PSMA–SMDC consisting of the high affinity targeting moiety DUPA, a disulfide linker and an indenoisoquinoline topoisomerase I inhibitor. Additionally, a peptide linker was used to improve the hydrophilicity and the orientation of the SMDC within the PSMA binding pocket. The DUPA–drug conjugate displayed high binding affinity in vitro along with antitumor efficacy and good tolerability in xenograft models. Based on these promising results, another DUPA–SMDC was developed using paclitaxel as the cytotoxic drug in addition to a disulfide linker to ensure tumor specific release. DUPA–PTX also achieved encouraging results in cell assays and animal studies [
27]. In 2019, a research group, from the company Endocyte, published preclinical results for EC1169, the first PSMA–SMDC to enter clinical trials. EC1169 is composed of the KuE–PSMA binding unit, the antimitotic drug tubulysin B hydrazide and a disulfide-based linker. It showed outstanding features in terms of good binding affinity in vitro, and high therapeutic efficacy and safety in vivo compared to docetaxel, which is the standard of care chemotherapeutic agent in the management of mCRPC. This favorable profile encouraged further testing in a phase 1 clinical trial [
28].
Wang et al. presented the results obtained from preclinical studies of a novel PSMA targeting SMDC. PSMA-1–VcMMAE was developed, based on PSMA–ADC through replacement of the mAb by the small molecule PSMA-1. Both PSMA drug conjugates use MMAE as the cytotoxic drug and the cathepsin cleavable linker valine–citrulline. However, PSMA–ADC displayed higher cytotoxic potency in PSMA-positive cells, probably due to the higher affinity of the mAb to PSMA. Nevertheless, animal studies proved the superiority of PSMA-1–VcMMAE over PSMA–ADC in terms of a larger therapeutic index [
29]. A similar approach was applied by Boinapally et al. [
30], who recently reported the development of SBPD-1 which consists of MMAE, valine–citrulline linker and a small-molecule PSMA-binding unit. SBPD-1 demonstrated high binding affinity in the low-nanomolar range along with PSMA-dependent cytotoxicity and antitumor effect in xenograft models. The tolerability and, thus, the translational potential of this SMDC were the main highlighted benefits.
In summary, the herein described SMDCs (
Table 1) have to prove their translational potential by undergoing clinical testing. However, it will be exciting to see whether they succeed in circumventing the drawbacks of ADCs to, thus, avoid a similar fate.
Developing a small molecule drug conjugate requires a thorough design and the right choice of building components. The SMDC described in this study consists of three main modules. The targeting unit KuE-617 refers to the combination of the PSMA-inhibitor KuE (lysine-urea-glutamate) and the naphthyl-cyclohexyl linker of PSMA-617. The chemical structure of these compounds is shown in
Figure 1. This moiety is crucial in the design of targeted drug delivery systems since it is responsible for delivering the cytotoxic payload to the desired “address”. Obviously, this cytotoxic payload needs to be a highly potent drug, mostly with a narrow therapeutic window and, therefore, not applicable as a single drug. Herein, the potent antimitotic drug of choice was Monomethyl auristatin E (MMAE), which is one of several ADCs, such as the FDA-approved brentuximab vedotin (ADCETRIS™). Besides the essential components, the linker plays a decisive role in the efficacy, stability and resulting tolerability of the whole SMDC. In the developed SMDC, a valine–citrulline linker was used to conjugate MMAE to KuE-617. This dipeptide belongs to the class of enzyme-cleavable linkers. It is cleaved by cathepsin B, which is a lysosomal protease overexpressed in various forms of cancer, including prostate cancer [
31,
32,
33,
34].
3. Discussion
The development of strategies for selective drug delivery is one of the most important research fields in the fight against cancer. Herein, we described a small-molecule drug conjugate consisting of the potent antimitotic drug MMAE and the high affinity PSMA inhibitor derivative KuE-617. Both entities were linked via a valine–citrulline linker. The design, synthesis and subsequent in vitro and in vivo evaluations were performed in several steps.
The compound MMAE.VC.SA.617 was synthesized in a fast and straightforward two-step synthesis. The two units of the compound, the drug-linker conjugate MMAE.VC and the PSMA-binding unit KuE-617, were conjugated via asymmetric amidation using a squaric acid linking unit. In the first step, squaric acid had to be added every second day, as it was observed, via LC–MS, to be consumed during the reaction. After eight days, MMAE had been quantitatively converted. In this case, the amidation was carried out in an aqueous buffer solution, as the progress of the reaction was controlled by the pH value. However, since the MMAE conjugate did not dissolve completely in water, DMSO was added. The second stage of the reaction was carried out in ethanol. In the organic medium, triethylamine was added as a base to enable the second asymmetric amidation of the squaric acid linker unit. The final compound MMAE.VC.SA.617 was obtained after semi-preparative HPLC purification.
The PSMA-binding affinities of MMAE.VC.SA.617, as well as of the drug-free conjugate KuE-617, were determined in a cell-based radioligand competitive assay. The IC50 value of KuE-617 was in the low nanomolar range, similar to the chelator-based PSMA radioligands PSMA-617 and PSMA-11 (21.5 ± 1.9 nM, 15.1 ± 3.8 nM and 17.4 ± 1.6 nM respectively). However, the insertion of MMAE led to a significant decrease in PSMA-binding affinity resulting in a nine-fold higher IC50 value than that of KuE-617 (188.6 ± 24.7 nM vs. 21.5 ± 1.9 nM), indicating that the conjugation of KuE-617 to MMAE had a negative impact on the binding affinity of the PSMA inhibitor, probably due to changes in the conformation of the drug conjugate and its orientation within the PSMA binding pocket.
In the field of drug targeting, selecting a potent drug is as important as designing a high affinity targeting unit. The cytotoxic drug used herein was the tubulin inhibitor MMAE, which is already used in several ADCs [
38,
39,
40]. To characterize the pharmacological properties of the MMAE conjugate we determined its cytotoxicity using CellTiter-Blue
®. This assay is based on the ability of viable cells to transform resazurin into the fluorescence-emitter resorufin. Thus, the fluorescence signal correlates with cell viability. The IC
50 value of the single drug MMAE was, as expected, in the picomolar range, whereas the cytotoxicity of MMAE.VC.SA.617 was about 100-fold lower (0.23 ± 0.06 nM vs. 33.0 ± 4.9 nM). This could be related to a potential decrease in lipophilicity of MMAE.VC.SA.617 due to the added carboxylic groups of the KuE unit and the ureido group of citrulline, resulting in a reduction in passive diffusion through the cell membrane.
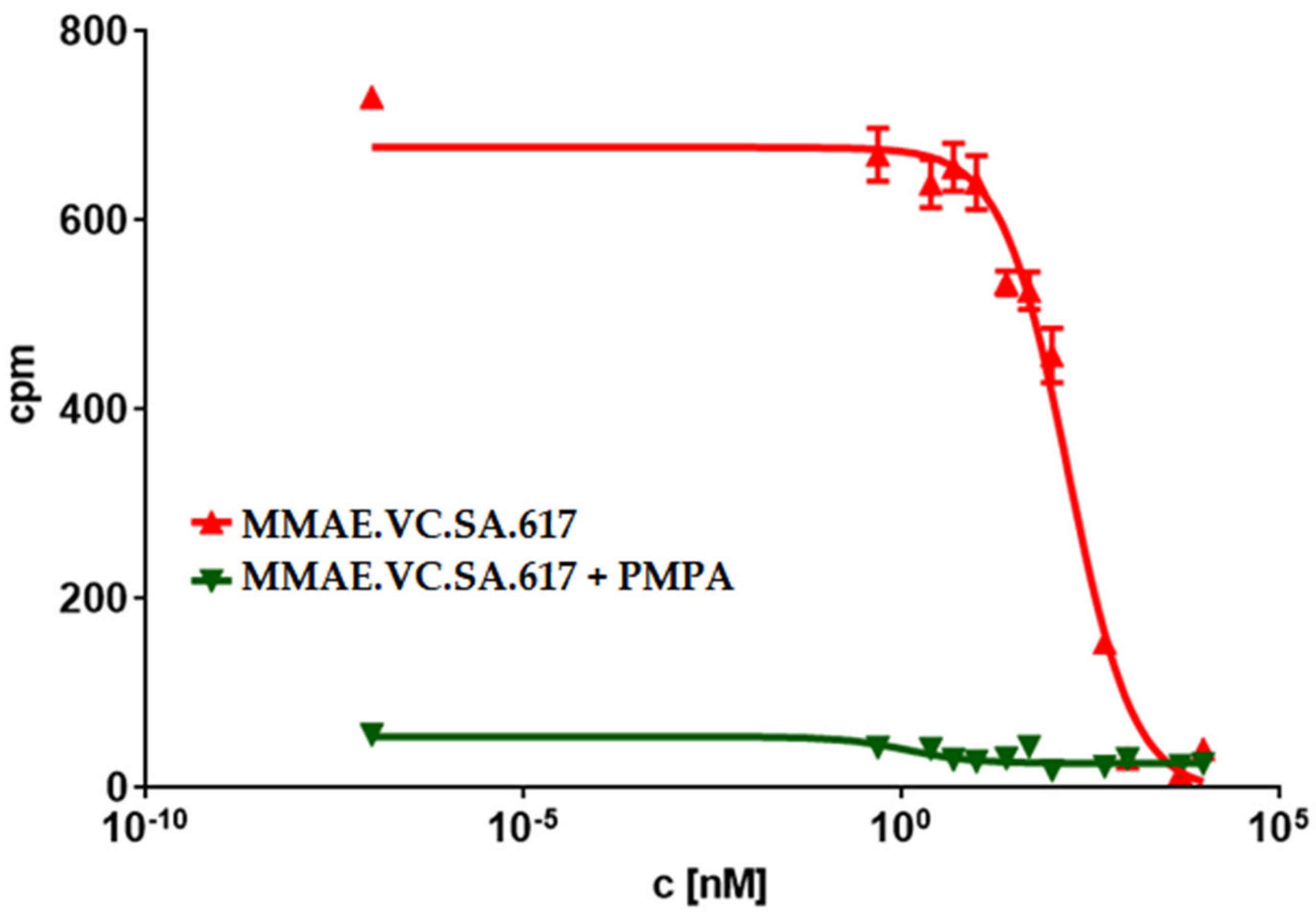
Another possible reason could be incomplete release of MMAE within tumor cells as a result of a low cathepsin B level in LNCaP cells or even an impaired internalization ratio. Nevertheless, the PSMA-specific uptake of MMAE.VC.SA.617 could be demonstrated by blocking PSMA receptors using PMPA which led to a three-times lower cytotoxicity (92.8 ± 8.3 nM). Additionally, the inhibition of cathepsin B via co-incubation with E-64 and the resulting decrease in cytotoxicity proved the essential role that this enzyme plays in the cleavage of the valine–citrulline linker and the subsequent release of MMAE. This enzyme-dependent cleavage of the dipeptide linker is a crucial feature in targeted therapeutics, since the active drug should be released only after uptake in tumor cells.
In a further step, we tried to further characterize the cytotoxicity of MMAE.VC.SA.617 using immuno-fluorescence imaging. It is known that the antimitotic drug MMAE acts by inhibiting the α-tubulin polymerization. This was proved with the conducted immunofluorescence studies, which showed a distinctive disturbance in tubulin formation of LNCaP cells incubated with either MMAE or MMAE.VC.SA.617 (
Figure 3b,c). The cytotoxic effect of MMAE.VC.SA.617 could be reduced by co-incubation with PMPA, which led to blocking of PSMA receptors (
Figure 3d). These results were in accordance with the findings described above.
The targeted delivery of cytotoxic drugs to tumor cells requires not only binding to tumor-associated structures but also a specific release of the conjugated drug in tumor tissue. The SMDC described herein included a valine–citrulline linker, which is one of the commonly used linkers in ADCs [
31,
32]. Valine–citrulline is cleaved by enzymes of the cathepsin family, especially cathepsin B, which is highly expressed in tumor cells [
33,
34]. In order to verify the cathepsin-specific cleavage of MMAE.VC.SA.617, the ratio of MMAE over time in the presence or absence of cathepsin B was quantified (
Figure 4a,b). As expected, MMAE was completely released after about 20 min of incubation with cathepsin B, whereas MMAE.VC.SA.617 remained almost stable in PBS.
Based on the positive results obtained from the in vitro assays, animal studies were performed in order to characterize the in vivo profile of MMAE.VC.SA.617 in terms of antitumor effect and tolerability. The single drug MMAE was used as the reference. According to the results from the initial toxicity study, three different concentrations of MMAE.VC.SA.617 were selected: 0.1 mg/kg corresponding to 0.05 mg MMAE, 0.5 mg/kg corresponding to 0.25 mg MMAE and 1 mg/kg MMAE.VC.SA.617, corresponding to 0.49 mg/kg MMAE. This MMAE concentration range was found to be well-tolerated by the mice during the initial toxicity study. Twelve days after inoculation of the mice with LNCaP tumor cells, treatment was initiated, according to
Figure 6 (shown above). All five treatment groups received therapy on days 0 (first day of treatment), 4, 7, 11, 14, 18, 25 and 32. The control group of mice received 0.9% NaCl on the same days.
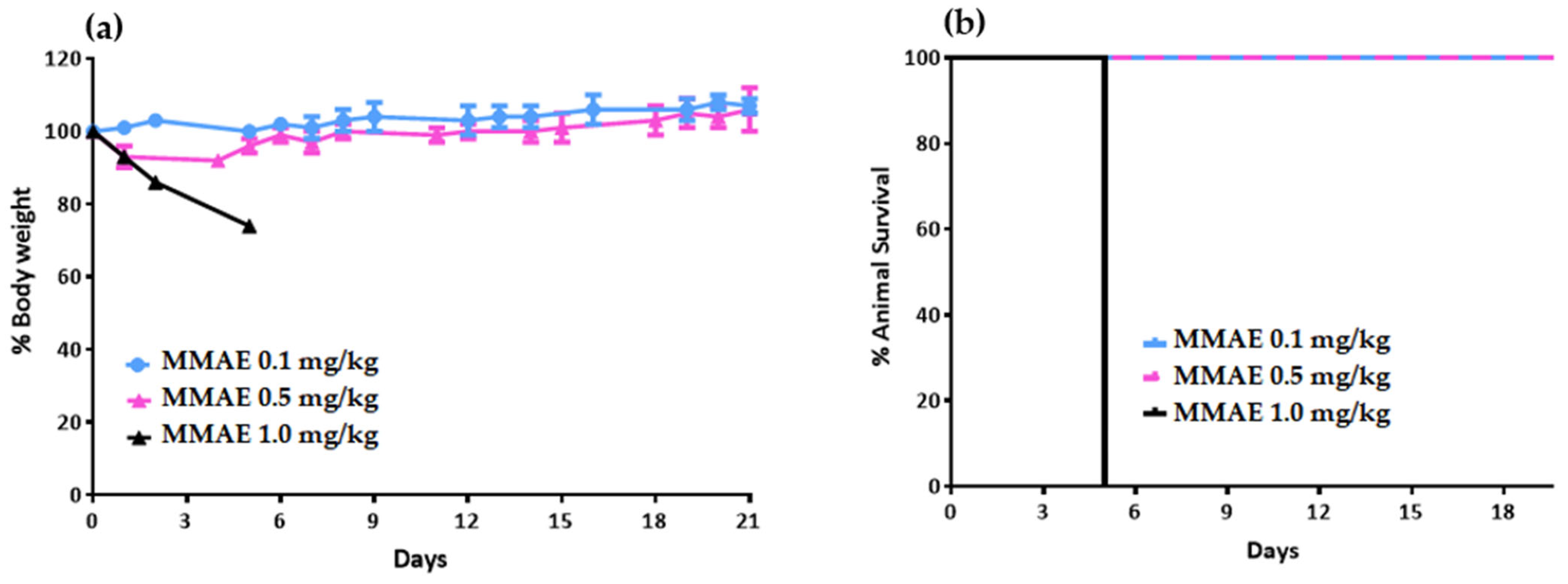
Animals treated with 0.1 mg/kg MMAE survived until day 51 of treatment and showed a constant tumor volume; however, the overall condition of the mice had deteriorated, and, thus, they had to be euthanized in accordance with bioethics principles (
Figure 7 and
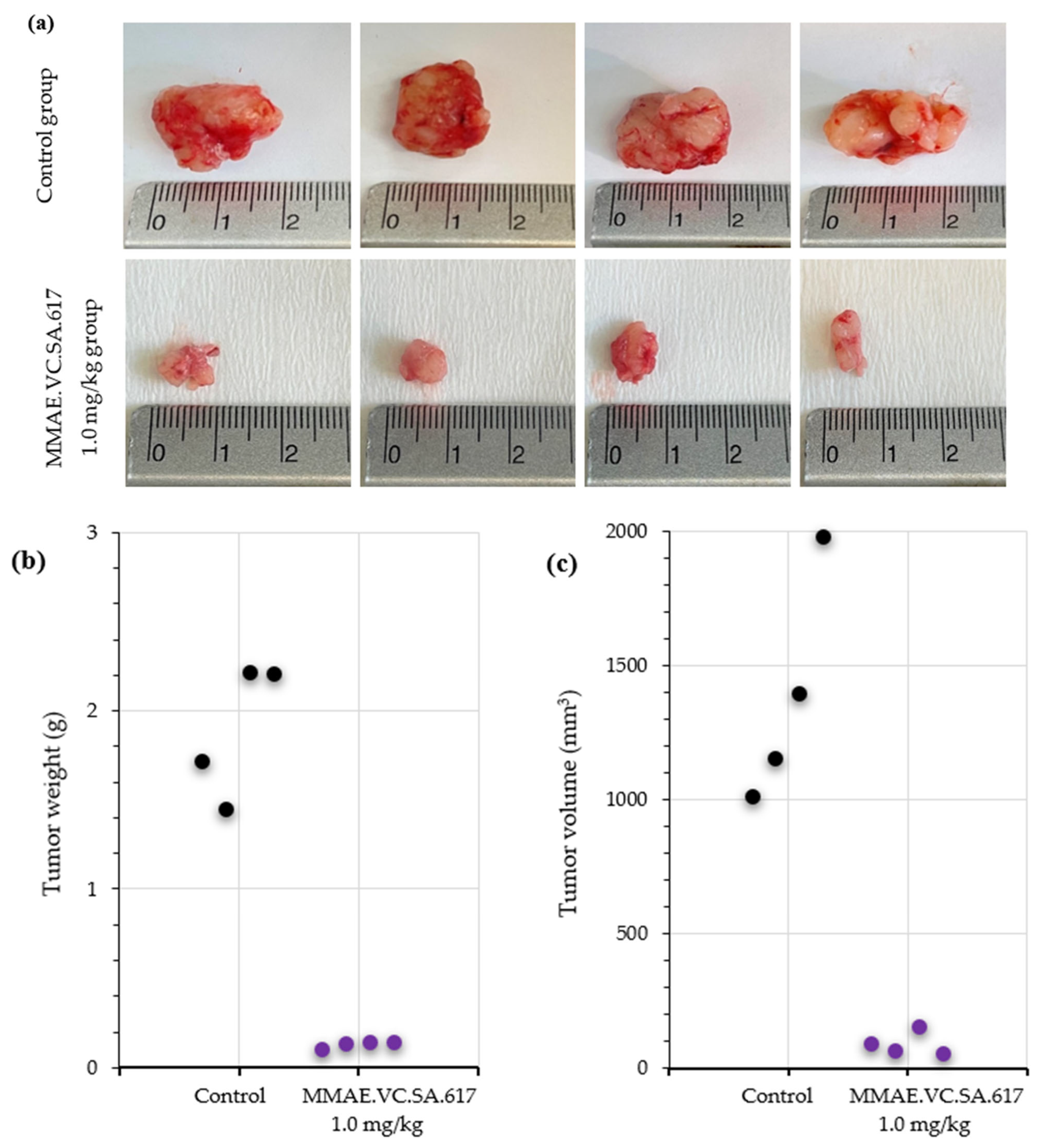
Figure 8). On the other hand, the mice in the 0.5 mg/kg MMAE group had to be euthanized by day 12 of the treatment due to approximately 20% weight loss, indicating severe toxicity of the single drug MMAE. The mice treated with 0.1 mg/kg MMAE.VC.SA.617 were euthanized before the end of the experiment due to body weight loss and large tumor volume. These results could be attributed to insufficient drug delivery into the tumor tissue. As concluded from in vitro studies the cytotoxicity of the MMAE conjugate was ten-fold lower in magnitude than the single drug MMAE, as shown above. This loss in cytotoxicity could not be compensated by active PSMA-targeting since MMAE.VC.SA.617 displayed moderate PSMA-binding affinity. Despite these results for the 0.1 mg/kg MMAE.VC.SA.617 group, tolerability to MMAE.VC.SA.617 of the two other concentrations (0.5 mg/kg and 1.0 mg/kg) was demonstrated (100% animal survival) even after eight intravenous injections. Moreover, tumor growth was effectively inhibited. The 0.5 mg/kg MMAE.VC.SA.617 treatment dose was tolerated well in terms of tumor volume and body weight; however, as in the case of the 0.1 mg/kg MMAE mice, the mice had to be euthanized due to poor body score. The mice which were administered with 1 mg/kg MMAE.VC.SA.617 showed impressive tumor regression after the second therapeutic administration (day 3 of therapy), which remained practically stable until the end of the experiment (day 63).
The analysis of the histopathologic features of the tumors revealed increased necrotic areas, mostly located in the central area of the tumors. The control tumors showed extensive necrotic areas, which were attributed to the lack of vascularization. On the contrary, both the treated and the control tumors showed normal proliferating rates on the periphery. However, treatment with MMAE.VC.SA.617 resulted in a six-fold higher apoptotic rate compared to the control group (3.002% vs. 0.579%), indicating an impact on either the apoptotic rate of the cancer cells or their proliferating status, or both. The smaller tumors collected from the treated animals indicated a robust inhibition of their growth, which could be explained either via direct killing of the cancer cells or via inhibition of their proliferation. These robust effects of the treatment might have been ameliorated at the end of the experiment, which was almost a month after the last injection of MMAE.VC.SA.617, and, thus, the proliferation and apoptosis that we observed reflected their current status and not the initial anti-tumoral function of the treatment.
Finally, MMAE.VC.SA.617 showed high binding affinity and cytotoxicity in vitro; in the nanomolar range for both. Although the single agent MMAE exhibited almost 100-fold higher cytotoxicity than the single drug in vitro, the SMDC clearly showed its superiority in vivo. Treatment with 0.5 mg/kg MMAE resulted in significant side effects, and animals had to be euthanized several days after intravenous injection. In contrast, mice treated with the same amount of MMAE, this time as SMDC, showed tumor regression after the second injection, even over the entire experimental period of more than 60 days, with a survival rate of 100%.
In general, targeting PSMA by small molecules has been shown to be superior to the use of antibodies. All PSMA ADCs investigated to date, such as MLN2704, PSMA-ADC or MEDI3726, failed in clinical trials due to serious adverse effects resulting from premature drug release [
21,
22,
23,
24,
25,
29]. Inspired by the successful implementation of small molecule PSMA radiopharmaceuticals, several PSMA SMDCs were developed. EC1169 is probably the best-known member of this group, as it was the first to be investigated in clinical trials. EC1169, which is composed of the KuE–PSMA binding unit, the antimitotic drug tubulysin B hydrazide and a disulfide-based linker, showed outstanding features in terms of good binding affinity in vitro, high therapeutic efficacy and safety in vivo [
28,
41]. However, the results of phase 1 studies results did not reveal sufficient efficacy. PSMA–SMDC and DUPA–PTX are two other PSMA drug conjugates containing a disulfide linker. Although both compounds showed good results in vitro and in vivo, they failed to confer any advantage over therapy with the unconjugated drug [
26,
27]. These results might indicate that the use of the disulfide linker does not lead to the desired specific drug release and, thus, does not achieve the expected efficacy, especially with regard to the positive results of the two valine–citrulline based SMDCs, PSMA-1–VcMMAE and SBPD-1. Both conjugates showed promising properties in terms of in vitro binding affinity and antitumor effect in xenograft models [
29,
30].
Thus, in accordance with our findings, the use of MMAE as the cytotoxic payload and valine-citrulline as the linker was shown to be beneficial in terms of efficacy and tolerability. However, direct comparability between MMAE.VC.SA.617 and the reported compounds cannot be made, since the in vitro and in vivo studies conducted with both PSMA-1VcMMAE and SBPD-1 were based on the use of PC3 PIP cells. This cell line was transduced to overexpress PSMA to a significantly higher extent than the patient-derived LNCaP cells used for testing of MMAE.VC.SA.617 [
42].
4. Materials and Methods
4.1. General
Chemicals were purchased from Sigma-Aldrich, Merck, Darmstadt, Germany, VWR, AcrosOrganics and TCI, Darmastadt, Germany. MMAE.VC was purchased from Hycultec GmbH, Beutelsbach, Germany and PSMA-617-NH2 from Huayi Isotopes Co. Haiyu town Jiangsu, China. Deuterated solvents for NMR spectra were acquired from Deutero GmbH, Kastellaun, Germany. Silica gel 60 F254 coated aluminum plates from Merck, Darmstadt, Germany were used for thin layer chromatography. NMR measurements were performed on an Avance III 600 spectrometer (600 MHz, 5 mm TCI CryoProbe sample head with z-Gradient and ATM and SampleXPress Lite 16 sample changer) from Bruker, Billerica, MA, USA. The LC/MS measurements were performed on an Agilent Technologies 1220 Infinity LC system coupled to an Agilent Technologies 6130B Single Quadrupole LC/MS system, Agilent Technologies GmbH, Waldbronn, Germany. Semi-preparative HPLC purification was performed on a 7000 series Hitachi LaChrom, Krefeld, Germany, using a semi-preparative LiChrospher 100 RP18 EC (250 × 10 mm) 5 µm column.
4.2. Organic Synthesis
4.2.1. MMAE.VC.SA
MMAE.VC (20 mg, 0.018 mmol) was dissolved in 0.5 M phosphate buffer pH 7 (500 µL) and DMSO (500 µL). 3,4-Diethoxycyclobut-3-ene-1,2-dione (5 mg, 4 µL, 0.027 mmol) was added and stirred for 8 days. Every second day 0.5 eq of 3,4-Diethoxycyclobut-3-ene-1,2-dione was added. The solvent was removed via lyophilization and the product was used in the next step without purification.
MS (ESI+): 236.0 ([M + H]+/2), calculated for C64H98N10O15: 1246.72 [M]+.
4.2.2. MMAE.VC.SA.617
MMAE.VC.SA (20 mg, 0.016 mmol) and PSMA-617-NH2 (10 mg, 0.016 mmol) were dissolved in ethanol (3 mL). Triethylamine (50 µL) was added and the reaction mixture was stirred for 6 days. The solvent was removed under reduced pressure. MMAE.VC.SA.617 was obtained as a white powder (14.2 mg, 43%) after HPLC purification (LiChrospher 100 RP18 EC (250 × 10 mm) 5 μL, flow rate: 5 mL/min, H2O/MeCN + 0.1% TFA, 45% to 55% MeCN in 20 min, tR = 9.0 min).
MS (ESI+): 929.0 ([M + H]+/2), calculated for C95H137N15O23: 1857.22 [M]+.
1H NMR (600 MHz, EtOD-d6) δ [ppm] = 7.77 (dd, J = 19.3, 9.2 Hz, 2H), 7.69 (d, J = 31.8 Hz, 3H), 7.41 (t, J = 7.9 Hz, 5H), 7.30 (dt, J = 15.4, 7.4 Hz, 3H), 7.24–7.15 (m, 1H), 5.18 (dt, J = 28.3, 15.2 Hz, 1H), 5.07 (d, J = 11.7 Hz, 1H), 4.93 (d, J = 31.6 Hz, 1H), 4.32–4.04 (m, 4H), 3.93–3.87 (m, 1H), 3.84 (s, 1H), 3.70 (s, 3H), 3.45 (d, J = 14.1 Hz, 2H), 3.41–3.26 (m, 9H), 3.23 (s, 1H), 3.11 (d, J = 15.3 Hz, 3H), 2.96 (td, J = 18.7, 7.0 Hz, 3H), 2.52 (s, 1H), 2.34–2.15 (m, 3H), 2.06 (s, 1H), 2.01–1.77 (m, 2H), 1.75–1.27 (m, 12H), 1.27–1.16 (m, 6H), 1.08–0.69 (m, 28H).
4.3. In Vitro Binding Affinity
LNCaP prostate cancer cells (purchased from Sigma-Aldrich, Darmstadt, Germany) were cultured in RPMI 1640 (Thermo Fisher Scientific, Dreieich, Germany) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), 100 μg/mL streptomycin, and 100 units/mL penicillin at 37 °C in 5% CO2.
LNCaP cells were incubated for 45 min with different concentrations of the MMAE-conjugates in the presence of 0.75 nM [68Ga]Ga-PSMA-10. Free radioactivity was removed by several washing steps with ice-cold PBS. Probes were measured in a γ-counter (2480 WIZARD2 Automatic Gamma Counter, PerkinElmer, Rodgau, Germany). Obtained data were analyzed in GraphPad Prism 9, GraphPad Software Boston, MA, USA, using nonlinear regression.
4.4. CellTiter-Blue® Viability Assay
A total of 104 cells per well were seeded in a 96-well plate for 24 h prior to incubation with increasing concentrations of either MMAE (0.1 nM to 0.5 µM) or MMAE.VC.SA.617 (2.5 nM to 10 µM). Subsequently, 20 µL of CellTiter-Blue® Reagent were added in each well and incubated for 2 h at 37 °C. For blocking studies, 2.5 nmol of PMPA was added to each well prior to incubation with SMDC. Fluorescence (560Ex/590Em) was recorded using a Tecan Spark multimode reader.
4.5. Immunofluorescence Studies
A total of 2000 cells/well were seeded in a Nunc® Lab-Tek® II—CC2™ Chamber Slide™ (Sigma Aldrich) and incubated with the test compounds at 37 °C for 24 h. After fixation with 4% PFA, cells were permeabilized with 0.5% Triton X-100 for 15 min at room temperature. Cells were then washed several times with PBS and blocked with 3% BSA in PBS for 1 h at room temperature. α-tubulin staining was performed by incubating the cells with alpha-Tubulin Antibody, Alexa Fluor® 488 conjugate (B-5-1-2) (Thermo Fisher Scientific) at a final concentration of 2 µg/mL for 3 h at room temperature. Counterstaining with DAPI was carried out with ProLong™ Gold Antifade Mountant with DAPI (Thermo Fisher Scientific), according to the manufacturer’s protocol. Cells were visualized using a fluorescence microscope (Keyence BZ-8000, KEYENCE DEUTSCHLAND GmbH, Neu-Isenburg, Germany) at 20×.
4.6. Cathepsin B Cleavage Assay
Cathepsin B from human liver (Sigma Aldrich) was activated by incubation at room temperature with 30 mM dithiothreitol DTT and 15 mM EDTA at pH 5.5. Subsequently, 2.5 µM of the activated cathepsin B was added to 25 µM of MMAE.VC.SA.617 and incubated at 37 °C. Aliquots were taken at different time points. The enzymatic activity of cathepsin B was blocked by adding 1 µL of E-64 (1 mM) in each vial. Samples were analyzed using an Agilent Technologies 1220 Infinity LC system coupled to an Agilent Technologies 6130B Single Quadrupole LC/MS system.
4.7. Animal Studies
Animals used for the biodistribution studies were obtained from the breeding facilities of the Institute of Biosciences and Applications, NCSR “Demokritos”. This experimental animal facility is registered according to the Greek Presidential Decree 56/2013 (Reg. Number: EL 25 BIO 022), in accordance with the European Directive 2010/63, which is harmonized with national legislation, on the protection of animals used for scientific purposes. All applicable national guidelines for the care and use of animals were followed. The study protocol was approved by the Department of Agriculture and Veterinary Service of the Prefecture of Athens.
4.7.1. Toxicology Study in Healthy NOD/SCID Mice
Prior to the therapeutic efficacy study in LNCaP tumor-bearing mice, a toxicology study was carried out on healthy NOD/SCID mice to determine the tolerable dose of the single drug MMAE. Nine NOD/SCID mice were divided into 3 groups and received a single intravenous (i.v.) dose of MMAE. Group A received 0.1 mg/kg body weight. Group B received 0.5 mg/kg body weight, while Group C received 1 mg/kg body weight). Mice injected with 1 mg/kg MMAE had to be euthanized 5 days post-injection due to body-weight loss.
4.7.2. Therapeutic Efficacy Study in LNCaP Tumor-Bearing NOD/SCID Mice
A therapeutic efficacy study of the MMAE.VC.SA.617 vs. MMAE was performed on six groups of LNCaP tumor-bearing NOD/SCID mice, two of which acted as reference groups, while an additional group acted as the control group. LNCaP cells were cultured in RPMI-1640 medium of pH 7.4, supplemented with 10% FBS, 100 U/mL of penicillin, 100 μg/mL of streptomycin, 2 mM glutamine, 10 mM HEPES and 1 mM sodium pyruvate. Cell cultures were maintained in 75 cm2 flasks, grown at 37 °C in 5% CO2 in a humidified atmosphere and the medium was changed approximately every 72 h (cell doubling time is about 40 h). Cells in the exponential phase of their growth were harvested by a 10 min treatment with a 0.05% trypsin–0.02% EDTA solution and neutralized with medium containing serum immediately. Cultures at passages 8–10 were used for the experiments. For the LNCaP xenograft development, cells were suspended in 100 μL in RPMI-1640 medium (supplemented as described above) and 100 μL Matrigel (medium: Matrigel ratio 1:1) (1 × 106 cells/200 μL) and maintained on ice until the inoculation. All equipment (syringes and needles) was chilled on ice prior to use in tumor cell inoculation. The mice were subcutaneously inoculated under the left shoulder with the LNCaP cells. The animals were ready for experimentation approximately 14 days after cell inoculation, when the tumor reached a volume of about 300 mm3. Mice were randomly divided into six groups, as follows:
Group A: MMAE 0.1 mg/kg body weight (reference group A)
Group B: MMAE 0.5 mg/kg body weight (reference group B)
Group C: MMAE.VC.SA.617, 0.1 mg/kg body weight
Group D: MMAE.VC.SA.617, 0.5 mg/kg body weight
Group E: MMAE.VC.SA.617, 1.0 mg/kg body weight
Group F: Saline (control group)
All groups of mice were intravenously injected twice a week, for the first 5 doses of MMAE, MMAE.VC.SA.617 or saline, and then an additional 3 doses once a week, resulting in a total of 8 doses over a period of 7 weeks (100 μL injected volume per intravenous injection). Body weight and tumor volume were assessed on each day of intravenous injection, and every 3–4 days after the end of treatment administration, up to 63 days after the initiation of the therapeutic efficacy study (only the mice of Group E were monitored until day 63). Tumor volume was measured using calipers, and was calculated using the formula (length × width
2)/2 [
43,
44]. The tumor growth index (TGI) for both animal groups was calculated by dividing the tumor volume measured each day by the initial tumor volume on day 0, before initiation of treatment. TGI was plotted vs. treatment time post-injection.
4.7.3. Histology and Immunohistochemistry Staining
The tumors were fixed in 10% neutral buffered formalin (NBF, Sigma) and then routinely processed and paraffin embedded. Tumor sections were dewaxed and rehydrated and were then stained with hematoxylin and eosin (H&E). For immunohistochemistry, sections were antigen-retrieved with heat-induced or enzymatic method. Peroxidase activity was blocked using 1.5% hydrogen peroxide. Sections were blocked with different blocking protocols, depending on the antibody. Staining was performed using the following anti–mouse antibodies: anti-Ki67 (Cell Signaling, Danvers, MA, 01923 USA, REF 9449) (1:1000 dilution) and anti-Caspase 3 (Cell Signaling, 9661) (1:800 dilution). A polymer-based detection kit, which consisted of horseradish peroxidase–conjugated polymers was used for the detection. To determine proliferation indices, Ki67-positive and Ki67-negative cells were counted using ImageJ software (
https://imagej.net/software/fiji/downloads) in 8–10 representative fields of all the tumors (on average, ~3000 nuclei were counted per specimen). A similar approach was followed in order to evaluate the % percentage of apoptotic cells.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}