
Polymorphisms within Autophagy-Related Genes as Susceptibility Biomarkers for Multiple Myeloma: A Meta-Analysis of Three Large Cohorts and Functional Characterization
 , ,
, ,  , , , , , , ,
, , , , , , ,  , , , , ,
, , , , ,  ,
,  ,
,  , , , ,
, , , ,  , ,
, ,  ,
,  ,
,  and
and  add
Show full author list
add
Show full author list
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Association of Autophagy-Related Polymorphisms with the Risk of Developing MM
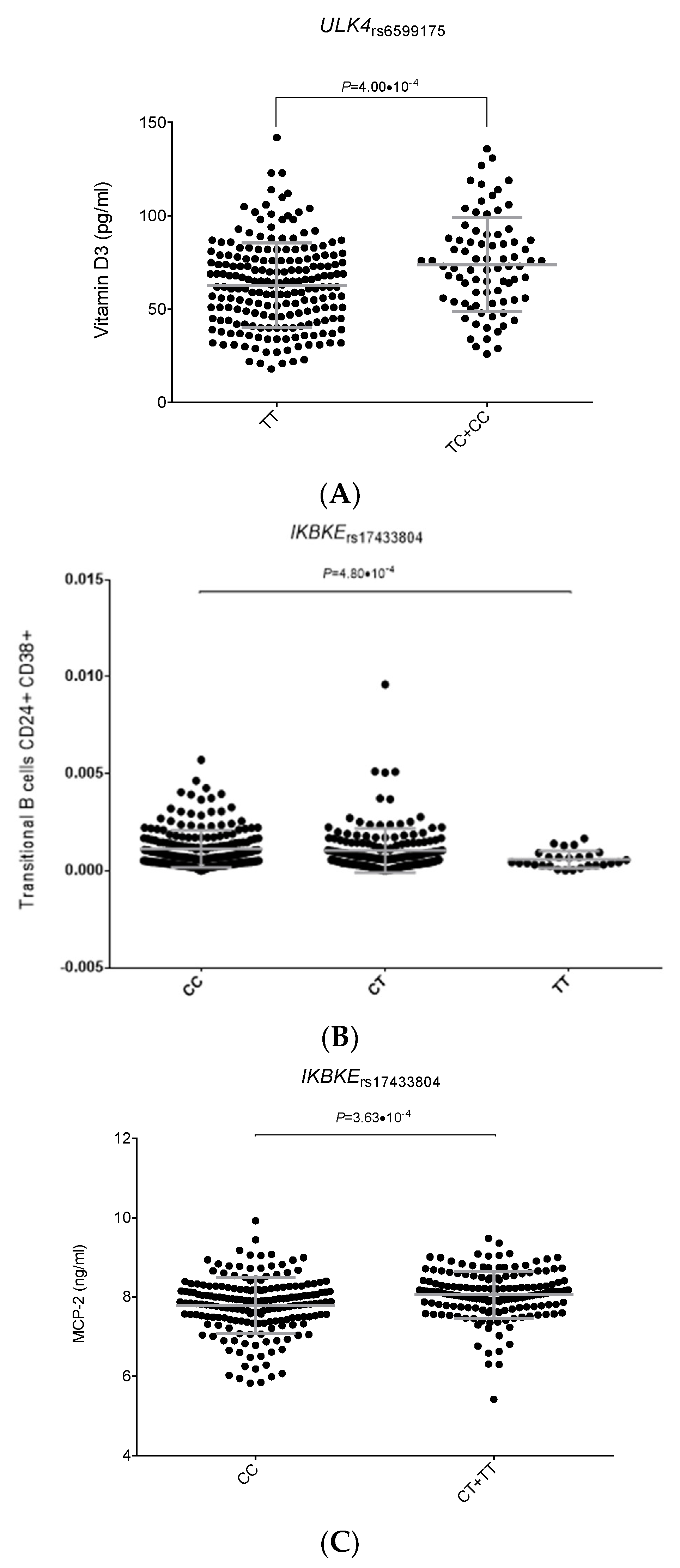
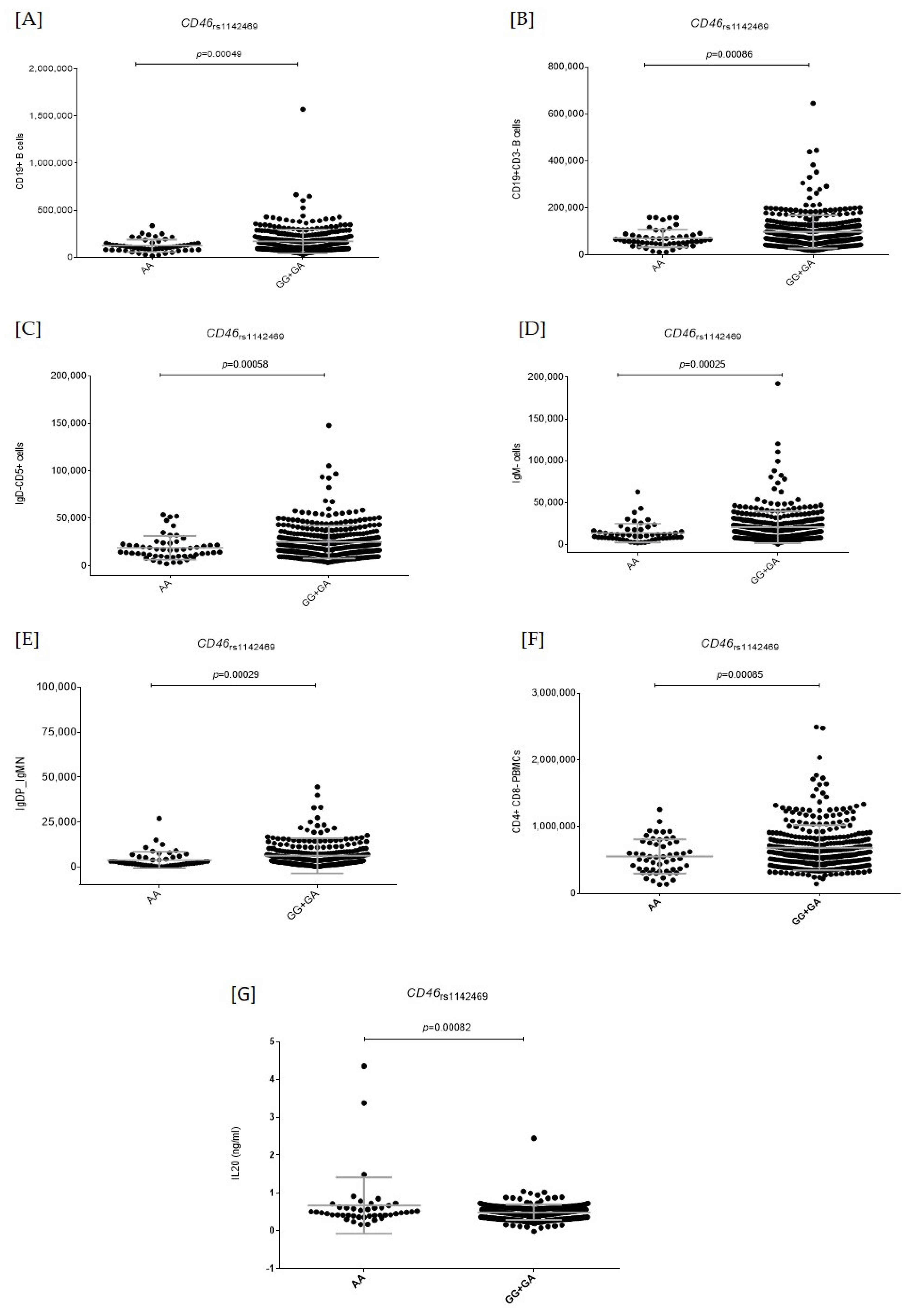
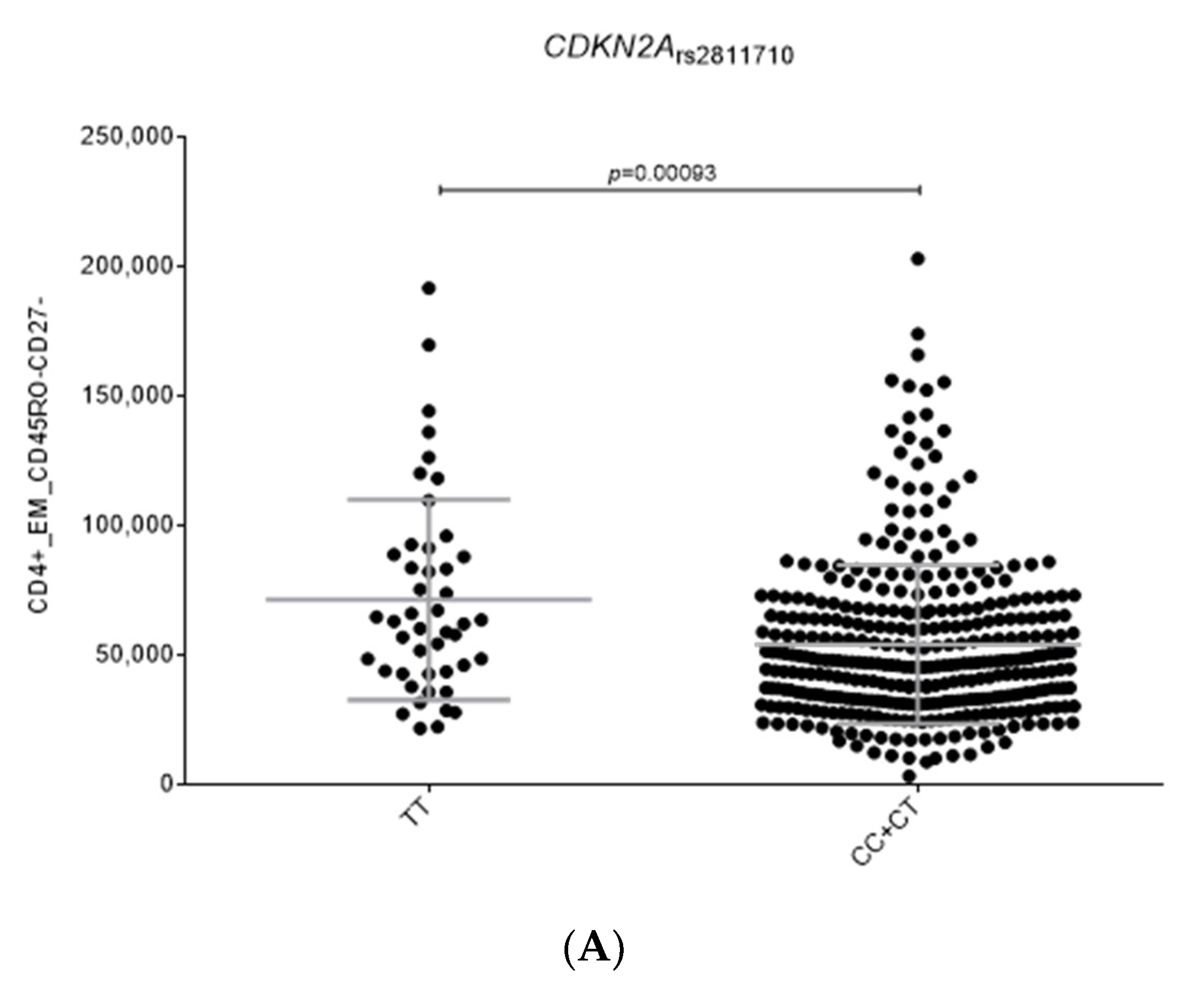
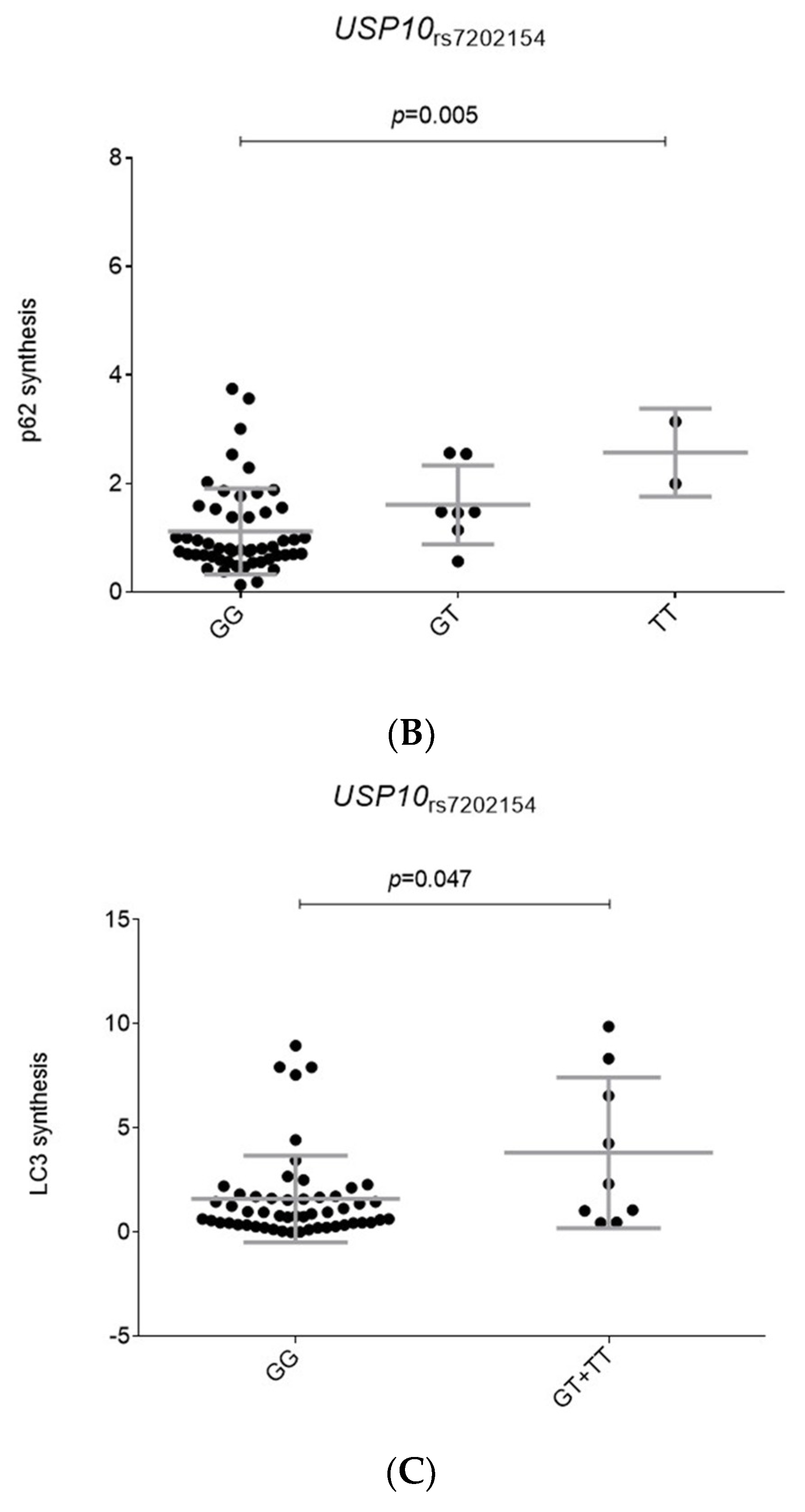
2.2. Functional Relevance of Autophagy-Related SNPs
3. Discussion
4. Materials and Methods
4.1. Study Populations
4.2. SNP Selection
4.3. Replication Cohort, Genotyping, and Meta-Analysis
4.4. Functional Effect of the Autophagy-Related Variants on Immune Responses
4.5. Correlation between Autophagy-Related SNPs and Steroid Hormone Levels
4.6. Impact of Autophagy-Related Variants on the Autophagy Flux
4.7. In Silico Functional Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, E.; Zagouri, F.; Symeonidis, A.; Roussou, M.; Sioni, A.; Pouli, A.; Delimpasi, S.; Katodritou, E.; Michalis, E.; Michael, M.; et al. Preserved levels of uninvolved immunoglobulins are independently associated with favorable outcome in patients with symptomatic multiple myeloma. Leukemia 2014, 28, 2075–2079. [Google Scholar] [CrossRef] [PubMed]
- Bradwell, A.; Harding, S.; Fourrier, N.; Mathiot, C.; Attal, M.; Moreau, P.; Harousseau, J.L.; Avet-Loiseau, H. Prognostic utility of intact immunoglobulin Ig’kappa/Ig’lambda ratios in multiple myeloma patients. Leukemia 2013, 27, 202–207. [Google Scholar] [CrossRef]
- Hoang, B.; Benavides, A.; Shi, Y.; Frost, P.; Lichtenstein, A. Effect of autophagy on multiple myeloma cell viability. Mol. Cancer Ther. 2009, 8, 1974–1984. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Landowski, T.H.; Megli, C.J.; Nullmeyer, K.D.; Lynch, R.M.; Dorr, R.T. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005, 65, 3828–3836. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Kung, A.L.; Davies, F.E.; Morgan, G.; Akiyama, M.; Shringarpure, R.; Munshi, N.C.; et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood 2006, 107, 1092–1100. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Arsov, I.; Adebayo, A.; Kucerova-Levisohn, M.; Haye, J.; MacNeil, M.; Papavasiliou, F.N.; Yue, Z.; Ortiz, B.D. A role for autophagic protein beclin 1 early in lymphocyte development. J. Immunol. 2011, 186, 2201–2209. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef]
- Schwarten, M.; Mohrluder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the immune system. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy as an innate immunity paradigm: Expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 2012, 24, 21–31. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy: An emerging immunological paradigm. J. Immunol. 2012, 189, 15–20. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; He, Y.W. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J. Immunol. 2011, 186, 5313–5322. [Google Scholar] [CrossRef] [PubMed]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef]
- Dykstra, K.M.; Allen, C.; Born, E.J.; Tong, H.; Holstein, S.A. Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget 2015, 6, 41535–41549. [Google Scholar] [CrossRef]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Mariggio, M.A.; Racanelli, V.; Vacca, A.; Frassanito, M.A. Autophagy: A New Mechanism of Prosurvival and Drug Resistance in Multiple Myeloma. Transl. Oncol. 2018, 11, 1350–1357. [Google Scholar] [CrossRef]
- Escalante, A.M.; McGrath, R.T.; Karolak, M.R.; Dorr, R.T.; Lynch, R.M.; Landowski, T.H. Preventing the autophagic survival response by inhibition of calpain enhances the cytotoxic activity of bortezomib in vitro and in vivo. Cancer Chemother. Pharmacol. 2013, 71, 1567–1576. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Tucci, M.; Stucci, S.; Savonarola, A.; Resta, L.; Cives, M.; Rossi, R.; Silvestris, F. An imbalance between Beclin-1 and p62 expression promotes the proliferation of myeloma cells through autophagy regulation. Exp. Hematol. 2014, 42, 897–908.e1. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Liang, C.; Jung, J.U. Autophagy genes as tumor suppressors. Curr. Opin. Cell Biol. 2010, 22, 226–233. [Google Scholar] [CrossRef]
- Huang, X.; Cao, W.; Yao, S.; Chen, J.; Liu, Y.; Qu, J.; Li, Y.; Han, X.; He, J.; Huang, H.; et al. NEDD4L binds the proteasome and promotes autophagy and bortezomib sensitivity in multiple myeloma. Cell Death Dis. 2022, 13, 197. [Google Scholar] [CrossRef]
- Jaganathan, S.; Malek, E.; Vallabhapurapu, S.; Vallabhapurapu, S.; Driscoll, J.J. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014, 5, 12358–12370. [Google Scholar] [CrossRef][Green Version]
- Di Lernia, G.; Leone, P.; Solimando, A.G.; Buonavoglia, A.; Saltarella, I.; Ria, R.; Ditonno, P.; Silvestris, N.; Crudele, L.; Vacca, A.; et al. Bortezomib Treatment Modulates Autophagy in Multiple Myeloma. J. Clin. Med. 2020, 9, 552. [Google Scholar] [CrossRef]
- Cheng, Y.; Qi, F.; Li, L.; Qin, Z.; Li, X.; Wang, X. Autophagy-related genes are potential diagnostic and prognostic biomarkers in prostate cancer. Transl. Androl. Urol. 2020, 9, 2616–2628. [Google Scholar] [CrossRef]
- Wang, L.; Fang, D.; Liu, Y. Autophagy-related genes are potential diagnostic biomarkers for dermatomyositis. Ann. Transl. Med. 2022, 10, 228. [Google Scholar] [CrossRef]
- Zhu, F.X.; Wang, X.T.; Zeng, H.Q.; Yin, Z.H.; Ye, Z.Z. A predicted risk score based on the expression of 16 autophagy-related genes for multiple myeloma survival. Oncol. Lett. 2019, 18, 5310–5324. [Google Scholar] [CrossRef]
- Went, M.; Sud, A.; Forsti, A.; Halvarsson, B.M.; Weinhold, N.; Kimber, S.; van Duin, M.; Thorleifsson, G.; Holroyd, A.; Johnson, D.C.; et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun. 2018, 9, 3707, Correction in Nat. Commun. 2019, 10, 213. [Google Scholar] [CrossRef]
- Mitchell, J.S.; Li, N.; Weinhold, N.; Forsti, A.; Ali, M.; van Duin, M.; Thorleifsson, G.; Johnson, D.C.; Chen, B.; Halvarsson, B.M.; et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat. Commun. 2016, 7, 12050. [Google Scholar] [CrossRef]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedlander, M.R.; t Hoen, P.A.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Alexandrakis, M.G.; Pappa, C.A.; Kokonozaki, M.; Boula, A.; Vyzoukaki, R.; Staphylaki, D.; Papadopoulou, A.; Androulakis, N.; Tsirakis, G.; Sfiridaki, A. Circulating serum levels of IL-20 in multiple myeloma patients: Its significance in angiogenesis and disease activity. Med. Oncol. 2015, 32, 42. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef]
- Hudlebusch, H.R.; Lodahl, M.; Johnsen, H.E.; Rasmussen, T. Expression of HOXA genes in patients with multiple myeloma. Leuk. Lymphoma 2004, 45, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Chau, T.L.; Gioia, R.; Gatot, J.S.; Patrascu, F.; Carpentier, I.; Chapelle, J.P.; O’Neill, L.; Beyaert, R.; Piette, J.; Chariot, A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem. Sci. 2008, 33, 171–180. [Google Scholar] [CrossRef]
- Leich, E.; Weissbach, S.; Klein, H.U.; Grieb, T.; Pischimarov, J.; Stuhmer, T.; Chatterjee, M.; Steinbrunn, T.; Langer, C.; Eilers, M.; et al. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion- and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013, 3, e102. [Google Scholar] [CrossRef]
- Gilad, N.; Zukerman, H.; Pick, M.; Gatt, M.E. The role of CD24 in multiple myeloma tumorigenicity and effects of the microenvironment on its expression. Oncotarget 2019, 10, 5480–5491. [Google Scholar] [CrossRef]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef]
- Gross Even-Zohar, N.; Pick, M.; Hofstetter, L.; Shaulov, A.; Nachmias, B.; Lebel, E.; Gatt, M.E. CD24 Is a Prognostic Marker for Multiple Myeloma Progression and Survival. J. Clin. Med. 2022, 11, 2913. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef]
- Valkovic, T.; Babarovic, E.; Lucin, K.; Stifter, S.; Aralica, M.; Seili-Bekafigo, I.; Duletic-Nacinovic, A.; Jonjic, N. Plasma Levels of Monocyte Chemotactic Protein-1 Are Associated with Clinical Features and Angiogenesis in Patients with Multiple Myeloma. Biomed. Res. Int. 2016, 2016, 7870590. [Google Scholar] [CrossRef]
- Broek, I.V.; Asosingh, K.; Vanderkerken, K.; Straetmans, N.; Van Camp, B.; Van Riet, I. Chemokine receptor CCR2 is expressed by human multiple myeloma cells and mediates migration to bone marrow stromal cell-produced monocyte chemotactic proteins MCP-1, -2 and -3. Br. J. Cancer 2003, 88, 855–862. [Google Scholar] [CrossRef]
- Sukhdeo, K.; Mani, M.; Zhang, Y.; Dutta, J.; Yasui, H.; Rooney, M.D.; Carrasco, D.E.; Zheng, M.; He, H.; Tai, Y.T.; et al. Targeting the beta-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 7516–7521. [Google Scholar] [CrossRef]
- Gyory, I.; Fejer, G.; Ghosh, N.; Seto, E.; Wright, K.L. Identification of a functionally impaired positive regulatory domain I binding factor 1 transcription repressor in myeloma cell lines. J. Immunol. 2003, 170, 3125–3133. [Google Scholar] [CrossRef]
- Fan, F.; Podar, K. The Role of AP-1 Transcription Factors in Plasma Cell Biology and Multiple Myeloma Pathophysiology. Cancers 2021, 13, 2326. [Google Scholar] [CrossRef]
- Jovanovic, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef]
- Jin, Z.; Zhou, S.; Ye, H.; Jiang, S.; Yu, K.; Ma, Y. The mechanism of SP1/p300 complex promotes proliferation of multiple myeloma cells through regulating IQGAP1 transcription. Biomed. Pharmacother. 2019, 119, 109434. [Google Scholar] [CrossRef]
- Viziteu, E.; Grandmougin, C.; Goldschmidt, H.; Seckinger, A.; Hose, D.; Klein, B.; Moreaux, J. Chetomin, targeting HIF-1alpha/p300 complex, exhibits antitumour activity in multiple myeloma. Br. J. Cancer 2016, 114, 519–523. [Google Scholar] [CrossRef]
- Gong, Y.; Zack, T.I.; Morris, L.G.; Lin, K.; Hukkelhoven, E.; Raheja, R.; Tan, I.L.; Turcan, S.; Veeriah, S.; Meng, S.; et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 2014, 46, 588–594. [Google Scholar] [CrossRef]
- Tanaka, A. Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Lett. 2010, 584, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Kay, D.M.; Stevens, C.F.; Hamza, T.H.; Montimurro, J.S.; Zabetian, C.P.; Factor, S.A.; Samii, A.; Griffith, A.; Roberts, J.W.; Molho, E.S.; et al. A comprehensive analysis of deletions, multiplications, and copy number variations in PARK2. Neurology 2010, 75, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Broderick, P.; Chubb, D.; Johnson, D.C.; Weinhold, N.; Forsti, A.; Lloyd, A.; Olver, B.; Ma, Y.; Dobbins, S.E.; Walker, B.A.; et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat. Genet. 2011, 44, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Ehret, G.B.; Rice, K.; Verwoert, G.C.; Launer, L.J.; Dehghan, A.; Glazer, N.L.; Morrison, A.C.; Johnson, A.D.; Aspelund, T.; et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009, 41, 677–687. [Google Scholar] [CrossRef]
- Folkersen, L.; van’t Hooft, F.; Chernogubova, E.; Agardh, H.E.; Hansson, G.K.; Hedin, U.; Liska, J.; Syvanen, A.C.; Paulsson-Berne, G.; Franco-Cereceda, A.; et al. Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 365–373. [Google Scholar] [CrossRef]
- Cross, H.S.; Lipkin, M.; Kallay, E. Nutrients regulate the colonic vitamin D system in mice: Relevance for human colon malignancy. J. Nutr. 2006, 136, 561–564. [Google Scholar] [CrossRef]
- Pendas-Franco, N.; Aguilera, O.; Pereira, F.; Gonzalez-Sancho, J.M.; Munoz, A. Vitamin D and Wnt/beta-catenin pathway in colon cancer: Role and regulation of DICKKOPF genes. Anticancer Res. 2008, 28, 2613–2623. [Google Scholar]
- Rohan, J.N.; Weigel, N.L. 1Alpha,25-dihydroxyvitamin D3 reduces c-Myc expression, inhibiting proliferation and causing G1 accumulation in C4-2 prostate cancer cells. Endocrinology 2009, 150, 2046–2054. [Google Scholar] [CrossRef]
- Clay-Gilmour, A.I.; Hildebrandt, M.A.T.; Brown, E.E.; Hofmann, J.N.; Spinelli, J.J.; Giles, G.G.; Cozen, W.; Bhatti, P.; Wu, X.; Waller, R.G.; et al. Coinherited genetics of multiple myeloma and its precursor, monoclonal gammopathy of undetermined significance. Blood Adv. 2020, 4, 2789–2797. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Heuck, C.J.; Mehta, J.; Bhagat, T.; Gundabolu, K.; Yu, Y.; Khan, S.; Chrysofakis, G.; Schinke, C.; Tariman, J.; Vickrey, E.; et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J. Immunol. 2013, 190, 2966–2975. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, D.; Liu, L.; Stewart, A.K.; Berenson, J.R.; Lassam, N.; Hogg, D. Germline CDKN2A mutation implicated in predisposition to multiple myeloma. Blood 2000, 95, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Boyd, K.D.; Houlston, R.S.; Kaiser, M.F. Constitutional mutation in CDKN2A is associated with long term survivorship in multiple myeloma: A case report. BMC Cancer 2017, 17, 718. [Google Scholar] [CrossRef] [PubMed]
- Kay, N.E.; Leong, T.; Bone, N.; Kyle, R.A.; Greipp, P.R.; Van Ness, B.; Oken, M.M. T-helper phenotypes in the blood of myeloma patients on ECOG phase III trials E9486/E3A93. Br. J. Haematol. 1998, 100, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Zeller, T.; Wild, P.; Szymczak, S.; Rotival, M.; Schillert, A.; Castagne, R.; Maouche, S.; Germain, M.; Lackner, K.; Rossmann, H.; et al. Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PLoS ONE 2010, 5, e10693. [Google Scholar] [CrossRef] [PubMed]
- Rios-Tamayo, R.; Lupianez, C.B.; Campa, D.; Hielscher, T.; Weinhold, N.; Martinez-Lopez, J.; Jerez, A.; Landi, S.; Jamroziak, K.; Dumontet, C.; et al. A common variant within the HNF1B gene is associated with overall survival of multiple myeloma patients: Results from the IMMEnSE consortium and meta-analysis. Oncotarget 2016, 7, 59029–59048. [Google Scholar] [CrossRef]
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef]
- Martino, A.; Sainz, J.; Buda, G.; Jamroziak, K.; Reis, R.M.; Garcia-Sanz, R.; Jurado, M.; Rios, R.; Szemraj-Rogucka, Z.; Marques, H.; et al. Genetics and molecular epidemiology of multiple myeloma: The rationale for the IMMEnSE consortium (review). Int. J. Oncol. 2012, 40, 625–638. [Google Scholar] [CrossRef]
- Manuel Sanchez-Maldonado, J.; Martinez-Bueno, M.; Canhao, H.; Ter Horst, R.; Munoz-Pena, S.; Moniz-Diez, A.; Rodriguez-Ramos, A.; Escudero, A.; Sorensen, S.B.; Hetland, M.L.; et al. NFKB2 polymorphisms associate with the risk of developing rheumatoid arthritis and response to TNF inhibitors: Results from the REPAIR consortium. Sci. Rep. 2020, 10, 4316. [Google Scholar] [CrossRef]
- Li, Y.; Oosting, M.; Smeekens, S.P.; Jaeger, M.; Aguirre-Gamboa, R.; Le, K.T.T.; Deelen, P.; Ricano-Ponce, I.; Schoffelen, T.; Jansen, A.F.M.; et al. A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell 2016, 167, 1099–1110.e14. [Google Scholar] [CrossRef]
- Aguirre-Gamboa, R.; Joosten, I.; Urbano, P.C.M.; van der Molen, R.G.; van Rijssen, E.; van Cranenbroek, B.; Oosting, M.; Smeekens, S.; Jaeger, M.; Zorro, M.; et al. Differential Effects of Environmental and Genetic Factors on T and B Cell Immune Traits. Cell Rep. 2016, 17, 2474–2487. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| German GWAS (n = 3619) | InterLymph GWAS (n = 5100) | Meta-Analysis (n = 8719) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Gene | A1 | OR (95%CI) | p | OR (95%CI) | p | OR (95%CI) | p | PHet |
| rs2299864 | ATG5 | T | 1.18 (1.03–1.33) | 0.015 | 1.24 (1.12–1.36) | 1.96 × 10−5 | 1.22 (1.13–1.32) | 1.48 × 10−6 | 0.545 |
| rs1142469 | CD46 | A | 1.14 (1.02–1.25) | 0.020 | 1.11 (1.02–1.21) | 0.012 | 1.12 (1.05–1.20) | 9.68 × 10−4 | 0.694 |
| rs2811710 | CDKN2A | C | 1.15 (1.03–1.29) | 0.011 | 1.11 (1.02–1.20) | 0.0176 | 1.12 (1.05–1.20) | 5.28 × 10−4 | 0.623 |
| rs143309009 | CTSD | G | 2.22 (1.46–3.38) | 2.1 × 10−4 | 1.24 (0.89–1.72) | 0.2114 | 1.55 (1.20–2.01) | 9.14 × 10−4 | 0.032 |
| rs11064698 | HSPB8 | T | 1.53 (1.05–2.24) | 0.027 | 1.16 (1.06–1.27) | 0.0012 | 1.18 (1.08–1.28) | 2.46 × 10−4 | 0.163 |
| rs12739461 | IKBKE | T | 1.18 (1.05–1.32) | 0.0053 | 1.10 (1.01–1.20) | 0.028 | 1.13 (1.05–1.21) | 6.36 × 10−4 | 0.337 |
| rs2297546 | IKBKE | G | 1.22 (1.09–1.37) | 7.2 × 10−4 | 1.09 (1.01–1.18) | 0.037 | 1.13 (1.06–1.21) | 2.25 × 10−4 | 0.110 |
| rs17433804 | IKBKE | C | 1.24 (1.09–1.41) | 0.0011 | 1.08 (1.00–1.18) | 0.066 | 1.13 (1.05–1.21) | 7.51 × 10−4 | 0.086 |
| rs1884158 | PARK2 | T | 1.21 (1.08–1.35) | 0.00081 | 1.09 (1.00–1.18) | 0.049 | 1.14 (1.06–1.21) | 3.07 × 10−4 | 0.141 |
| rs34048269 | RPTOR | A | 1.30 (1.14–1.48) | 1.3 × 10−4 | 1.06 (0.97–1.17) | 0.199 | 1.14 (1.05–1.23) | 0.0024 | 0.013 |
| rs6599175 | ULK4 | C | 1.33 (1.16–1.53) | 5.7 × 10−5 | 1.25 (1.13–1.39) | 3.6 × 10−5 | 1.28 (1.18–1.39) | 4.00 × 10−9 | 0.482 |
| rs7202154 | USP10 | G | 1.31 (1.06–1.62) | 0.011 | 1.23 (1.05–1.44) | 0.0126 | 1.26 (1.11–1.43) | 3.83 × 10−4 | 0.640 |
| German GWAS (n = 3619) | InterLymph GWAS (n = 5100) | IMMEnSE (n = 3957) | Meta-Analysis (n = 12676) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene_SNP | OR (95%CI) | p | OR (95%CI) | p | OR (95%CI) | p | OR (95%CI) | PMeta | PHet |
| ATG5_rs2299864T | 1.18 (1.03–1.33) | 0.015 | 1.24 (1.12–1.36) | 1.96 × 10−5 | 1.07 (0.92–1.23) | 0.40 | 1.18 (1.11–1.27) | 1.3 × 10−6 Ϯ | 0.254 |
| CD46_rs1142469A | 1.14 (1.02–1.25) | 0.020 | 1.11 (1.02–1.21) | 0.012 | 1.09 (0.96–1.23) | 0.19 | 1.12 (1.05–1.18) | 2.2 × 10−4 | 0.851 |
| CDKN2A_rs2811710C | 1.15 (1.03–1.29) | 0.011 | 1.11 (1.02–1.20) | 0.0176 | 1.18 (1.06–1.32) | 0.003 | 1.14 (1.08–1.20) | 7.0 × 10−6 Ϯ | 0.666 |
| CTSD_rs143309009G | 2.22 (1.46–3.38) | 2.1 × 10−4 | 1.24 (0.89–1.72) | 0.2114 | 0.72 (0.47–1.10) | 0.13 | 1.26 (1.01–1.57) | 0.042 | 0.001 |
| HSPB8_rs11064698T | 1.53 (1.05–2.24) | 0.027 | 1.16 (1.06–1.27) | 0.0012 | 0.84 (0.72–0.99) | 0.035 | 1.09 (1.01–1.18) | 0.029 | 0.001 |
| IKBKE_rs12739461T | 1.18 (1.05–1.32) | 0.0053 | 1.10 (1.01–1.20) | 0.028 | 1.01 (0.90–1.14) | 0.88 | 1.10 (1.03–1.16) | 0.0022 | 0.179 |
| IKBKE_rs2297546G | 1.22 (1.09–1.37) | 7.2 × 10−4 | 1.09 (1.01–1.18) | 0.037 | 1.00 (0.90–1.12) | 0.94 | 1.10 (1.04–1.16) | 0.0013 | 0.047 |
| IKBKE_rs17433804C | 1.24 (1.09–1.41) | 0.0011 | 1.08 (1.00–1.18) | 0.066 | 1.16 (1.02–1.31) | 0.024 | 1.14 (1.07–1.21) | 4.6 × 10−5 Ϯ | 0.213 |
| PARK2_rs1884158T | 1.21 (1.08–1.35) | 0.00081 | 1.09 (1.00–1.18) | 0.049 | 1.04 (0.91–1.19) | 0.56 | 1.11 (1.05–1.18) | 4.5 × 10−4 | 0.184 |
| RPTOR_rs34048269A | 1.30 (1.14–1.48) | 1.3 × 10−4 | 1.06 (0.97–1.17) | 0.199 | 1.04 (0.91–1.19) | 0.56 | 1.11 (1.04–1.19) | 0.0017 | 0.024 |
| ULK4_rs6599175C | 1.33 (1.16–1.53) | 5.7 × 10−5 | 1.25 (1.13–1.39) | 3.6 × 10−5 | 1.37 (1.21–1.56) | 2.6 × 10−6 | 1.31 (1.22–1.40) | 5.8 × 10−14 Ϯ | 0.522 |
| USP10_rs7202154G | 1.31 (1.06–1.62) | 0.011 | 1.23 (1.05–1.44) | 0.0126 | 0.93 (0.76–1.15) | 0.50 | 1.16 (1.04–1.29) | 0.0075 | 0.048 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clavero, E.; Sanchez-Maldonado, J.M.; Macauda, A.; Ter Horst, R.; Sampaio-Marques, B.; Jurczyszyn, A.; Clay-Gilmour, A.; Stein, A.; Hildebrandt, M.A.T.; Weinhold, N.; et al. Polymorphisms within Autophagy-Related Genes as Susceptibility Biomarkers for Multiple Myeloma: A Meta-Analysis of Three Large Cohorts and Functional Characterization. Int. J. Mol. Sci. 2023, 24, 8500. https://doi.org/10.3390/ijms24108500
Clavero E, Sanchez-Maldonado JM, Macauda A, Ter Horst R, Sampaio-Marques B, Jurczyszyn A, Clay-Gilmour A, Stein A, Hildebrandt MAT, Weinhold N, et al. Polymorphisms within Autophagy-Related Genes as Susceptibility Biomarkers for Multiple Myeloma: A Meta-Analysis of Three Large Cohorts and Functional Characterization. International Journal of Molecular Sciences. 2023; 24(10):8500. https://doi.org/10.3390/ijms24108500
Chicago/Turabian StyleClavero, Esther, José Manuel Sanchez-Maldonado, Angelica Macauda, Rob Ter Horst, Belém Sampaio-Marques, Artur Jurczyszyn, Alyssa Clay-Gilmour, Angelika Stein, Michelle A. T. Hildebrandt, Niels Weinhold, and et al. 2023. "Polymorphisms within Autophagy-Related Genes as Susceptibility Biomarkers for Multiple Myeloma: A Meta-Analysis of Three Large Cohorts and Functional Characterization" International Journal of Molecular Sciences 24, no. 10: 8500. https://doi.org/10.3390/ijms24108500
APA StyleClavero, E., Sanchez-Maldonado, J. M., Macauda, A., Ter Horst, R., Sampaio-Marques, B., Jurczyszyn, A., Clay-Gilmour, A., Stein, A., Hildebrandt, M. A. T., Weinhold, N., Buda, G., García-Sanz, R., Tomczak, W., Vogel, U., Jerez, A., Zawirska, D., Wątek, M., Hofmann, J. N., Landi, S., ... Sainz, J. (2023). Polymorphisms within Autophagy-Related Genes as Susceptibility Biomarkers for Multiple Myeloma: A Meta-Analysis of Three Large Cohorts and Functional Characterization. International Journal of Molecular Sciences, 24(10), 8500. https://doi.org/10.3390/ijms24108500