
Strategies to Overcome Resistance to Immune-Based Therapies in Osteosarcoma

 ,
,  , , , ,
, , , ,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
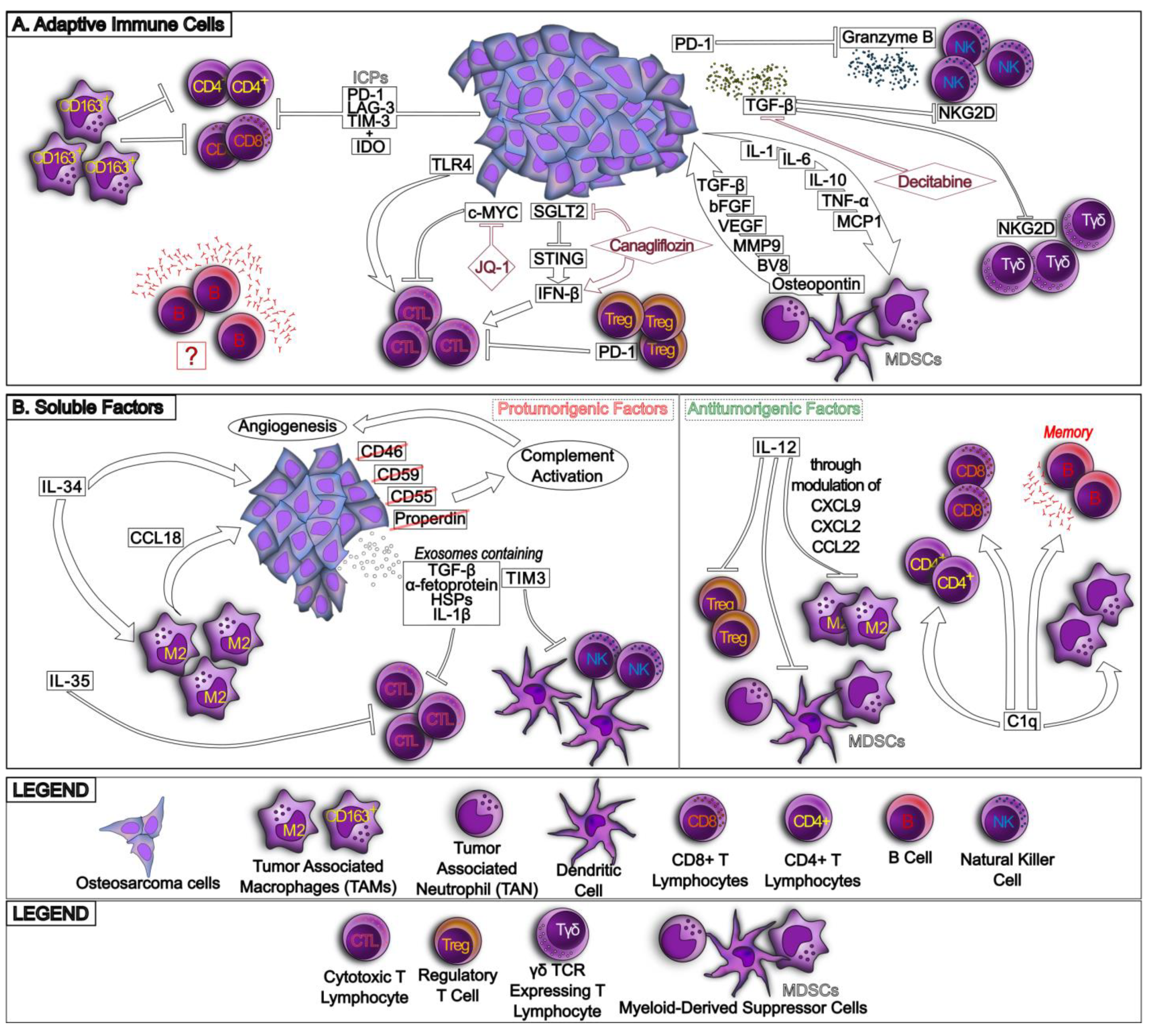
2. The Osteosarcoma Immune Environment
2.1. The Variegate Scenario of Osteosarcoma Immune Environment
2.2. Can the Immune Environment Characterization Be Used to Build Prognostic or Predictive Signatures?
3. Mechanisms of Resistance to the Host Immune System
3.1. Hypoxia Induces an Immunosuppressive Environment in Osteosarcoma
3.2. Resistance to Immunogenic Cell Death Induced by Chemotherapy in Osteosarcoma
4. Mechanisms of Resistance to Immunotherapy
4.1. Resistance to Immune Checkpoint Inhibitors
4.2. Resistance to CAR T Cells
4.3. How to Restore Immune System Activity and Immunotherapy Efficacy against Osteosarcoma: Biological Bases
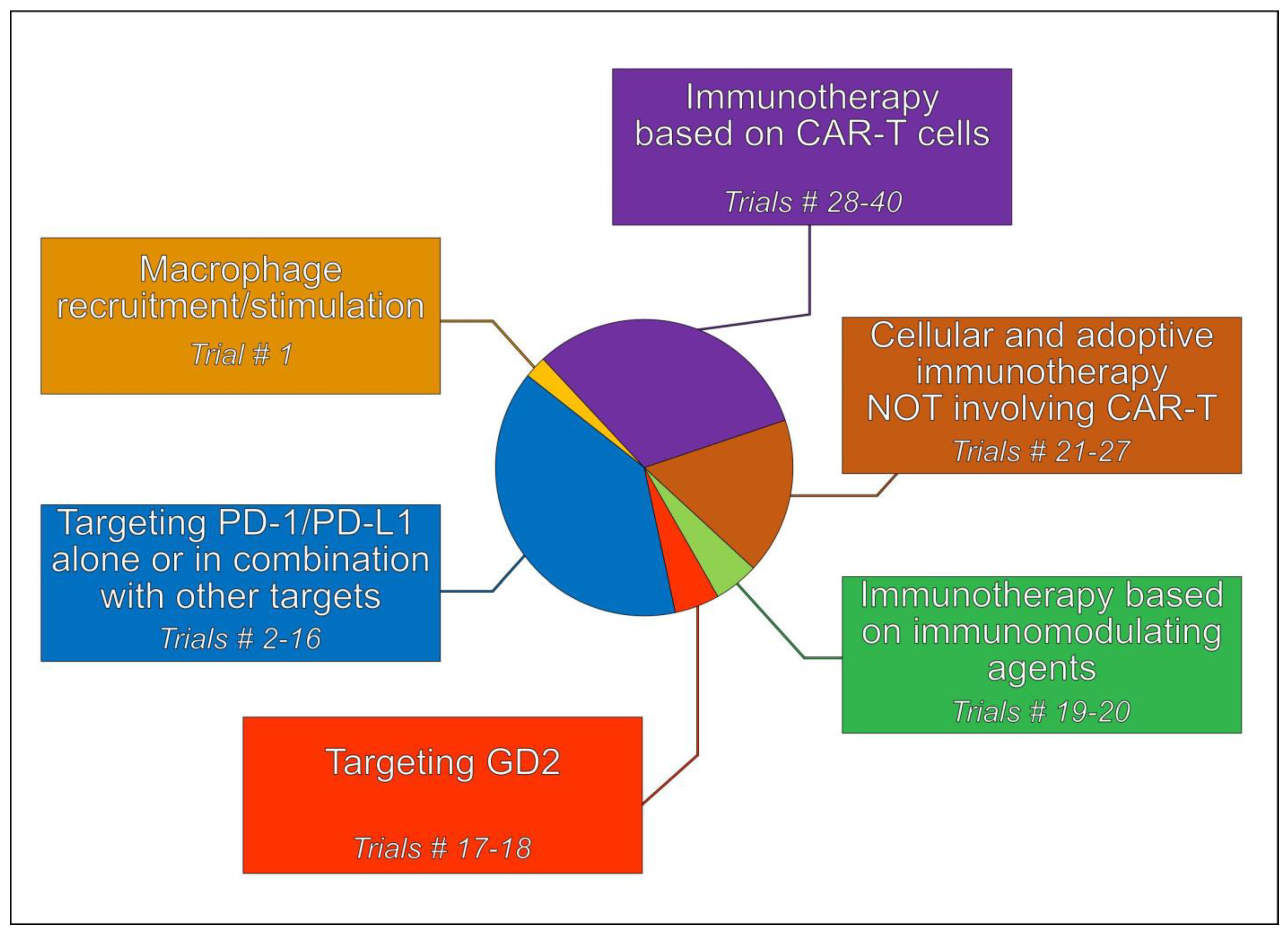
5. Immunotherapeutic Treatment Strategies
5.1. Tumor-Associated Macrophages (TAMs)
5.2. Immune Checkpoint Inhibitors (ICIs)
5.3. Targeting Disialoganglioside 2 (GD2)
5.4. Immunotherapy Based on Immunomodulating Agents
5.5. Cellular and Adoptive Immunotherapy Not Involving CAR T Cells
5.6. Immunotherapy Involving CAR T Cells
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABCA1 | ATP Binding Cassette Subfamily A Member 1 |
| ABCB1 | ATP Binding Cassette Subfamily B Member 1 |
| ALCAM/CD166 | activated leukocyte cell adhesion molecule |
| ATG16L1 | autophagy related 16 like 1 gene |
| ATP6V0D1 | ATPase H+ Transporting V0 Subunit D1 |
| attIL12 | anchored and tumor-targeted |
| bFGF | basic fibroblast growth factor |
| B7-H3 or CD276 | CD276 molecule |
| BTK | Bruton’s tyrosine kinase |
| CA IX | carbonic anhydrase IX |
| CAR | chimeric antigen receptor |
| CCL5 | chemokine (C-C motif) ligand 5 |
| CCL18 | chemokine (C-C motif) ligand 18 |
| CCL22 | chemokine (C-C motif) ligand 22 |
| CCL28 | chemokine (C-C motif) ligand 28 |
| CCR7 | C-C chemokine receptor type 7 |
| circRNAs | circular RNAs |
| CIK | cytokine-induced killer cells |
| CSF-1R | colony stimulating factor 1-receptor |
| CTLA4 | Cytotoxic T-Lymphocyte Associated Protein 4 |
| CTLs | cytotoxic T lymphocytes |
| CXCL2 | C-X-C Motif Chemokine Ligand 2 |
| CXCL9 | C-X-C Motif Chemokine Ligand 9 |
| CXCL22 | C-X-C Motif Chemokine Ligand 22 |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| DCs | dendritic cells |
| DEGs | differentially expressed genes |
| EGF | epithelial growth factor |
| EGFR | epithelial growth factor receptor |
| EMT | epithelial mesenchymal transition |
| ER | endoplasmic reticulum |
| GD2 | disialoganglioside 2 |
| GLUT-1 | Glucose Transporter 1 |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| GTEx | Genotype-Tissue Expression |
| Her2 (ERBB2) | Erb-B2 Receptor Tyrosine Kinase 2 |
| HGOS | high-grade osteosarcoma |
| HIF-1α | hypoxia inducible factors-1α |
| HSF1 | Heat shock protein transcription factor 1 |
| Hsp70 | Heat shock protein family A |
| HSPA1L | Heat shock protein family A member 1 like |
| ICD | immunogenic cell death |
| ICIs | immune checkpoint inhibitors |
| ICP | immune checkpoint |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-β | interferon-β |
| IFN-γ | interferon-γ |
| IGF1R | Insulin Like Growth Factor 1 Receptor |
| ILs | interleukins |
| IPP | isopentenyl pyrophosphate |
| IRAKs | Interleukin 1 Receptor Associated Kinases |
| IRF3 | interferon regulatory factor 3 |
| IRG | inflammation regulatory gene |
| ITGAM | Integrin subunit α M |
| LAG-3 | lymphocyte activating 3 |
| LGALBP | L-galectin 3 soluble binding protein 3 |
| L-MTP-PE | liposomal muramyl tripeptide phosphatidylethanolamine |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractact protein 1 |
| MDSCs | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| miRNAs | micro RNAs |
| MMP-9 | Matrix Metallopeptidase 9 |
| mRNAs | messenger RNAs |
| MSR1 | macrophage scavenger receptor 1 |
| mTOR | mammalian target of Rapamycin |
| NFAT | nuclear factor of activated T cells |
| NK | natural killer |
| NKG2D (KLRK1) | Killer Cell Lectin Like Receptor K1 |
| OS | Overall Survival |
| PARP | poly ADP-ribose polymerase |
| PBMC | peripheral blood mononuclear cells |
| PCNA | Proliferating cell nuclear antigen |
| PD-1 | programmed death-1 |
| PD-1L, PD-L1 | programmed death-ligand 1 |
| PD-2L, PD-L2 | programmed death-ligand 2 |
| PDCD1LG2 | programmed cell death 1 ligand 2 |
| PDGF | platelet-derived growth factor |
| PDGFD | platelet-derived growth factor D |
| PDX | patient’s derived xenograft |
| PFS | progression-free survival |
| PI3K | phosphatidylinositide 3-kinase |
| PI3Kδ/γ | phosphatidyl inositol-3 kinase δ/γ |
| PREB | Prolactin Regulatory Element Binding |
| PSMA | Prostate-specific membrane antigen |
| RBPs | RNA-binding proteins |
| ROR1 | Receptor Tyrosine Kinase Like Orphan Receptor 1 |
| RTK | tyrosine kinase |
| S100A6 | S100 Calcium Binding Protein A6 |
| SEMA4D | semaphorin 4D |
| SGLT2 | Sodium-glucose transporter 2 |
| ssGSEA | single-cell gene enrichment set analysis |
| STC2 | Stanniocalcin 2 |
| STING | stimulator of interferon genes |
| TAMs | tumor-associated macrophages |
| TANs | tumor-associated neutrophils |
| TARGET | Therapeutically Applicable Research to Generate Effective Treatments |
| TGF-β2 | transforming growth factor-β2 |
| Th1 | T-helper 1 |
| TIGIT | T-cell immunoreceptor with Ig and ITIM domains |
| TILs | tumor-infiltrating lymphocytes |
| TIM-3 | mucin domain 3 |
| TIME | tumor immune microenvironment |
| TLR4 | Toll-like receptor 4 |
| TNF-α | tumor necrosis factor-α |
| TNFRSF9 | TNF Receptor Superfamily Member 9 |
| Treg | T-regulatory |
| TSPYL2 | testis-specific protein Y like 2 |
| TYROBP | transmembrane immune signaling adaptor TYROBP (protein tyrosine kinase binding protein) |
| UPR | unfolded protein response |
| VAV1 | Vav Guanine Nucleotide Exchange Factor 1 |
| VEGF | vascular endothelial growth factor |
| WGCNA | Weighted gene coexpression network analysis |
References
- Gill, J.; Gorlick, R. Advancing therapy for osteosarcoma. Nat. Rev. Clin. Oncol. 2021, 18, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Liang, X.; Ren, T.; Huang, Y.; Zhang, H.; Yu, Y.; Chen, C.; Wang, W.; Niu, J.; Lou, J.; et al. The role of tumor-associated macrophages in osteosarcoma progression—Therapeutic implications. Cell Oncol. 2021, 44, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Heymann, M.F.; Lezot, F.; Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Smit, J.W.A.; Netea, M.G. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. 2018, 413, 102–109. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, J.H.; Lin, Z.H.; Lv, H.Y.; Ye, Z.M.; Chen, Y.P.; Zhang, X.Y. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of osteosarcoma. Aging 2020, 12, 3486–3501. [Google Scholar] [CrossRef]
- Cortese, N.; Soldani, C.; Franceschini, B.; Barbagallo, M.; Marchesi, F.; Torzilli, G.; Donadon, M. Macrophages in Colorectal Cancer Liver Metastases. Cancers 2019, 11, 633. [Google Scholar] [CrossRef]
- Jeys, L.M.; Grimer, R.J.; Carter, S.R.; Tillman, R.M.; Abudu, A. Post operative infection and increased survival in osteosarcoma patients: Are they associated? Ann. Surg. Oncol. 2007, 14, 2887–2895. [Google Scholar] [CrossRef]
- Chen, Y.U.; Xu, S.F.; Xu, M.; Yu, X.C. Postoperative infection and survival in osteosarcoma patients: Reconsideration of immunotherapy for osteosarcoma. Mol. Clin. Oncol. 2015, 3, 495–500. [Google Scholar] [CrossRef]
- Zhu, T.; Han, J.; Yang, L.; Cai, Z.; Sun, W.; Hua, Y.; Xu, J. Immune Microenvironment in Osteosarcoma: Components, Therapeutic Strategies and Clinical Applications. Front. Immunol. 2022, 13, 907550. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhang, X.; Wu, Y.; Yang, Y. Inhibition of macrophage polarization prohibits growth of human osteosarcoma. Tumour Biol. 2014, 35, 7611–7616. [Google Scholar] [CrossRef]
- Yang, D.; Liu, K.; Fan, L.; Liang, W.; Xu, T.; Jiang, W.; Lu, H.; Jiang, J.; Wang, C.; Li, G.; et al. LncRNA RP11-361F15.2 promotes osteosarcoma tumorigenesis by inhibiting M2-Like polarization of tumor-associated macrophages of CPEB4. Cancer Lett. 2020, 473, 33–49. [Google Scholar] [CrossRef]
- Ren, S.; Zhang, X.; Hu, Y.; Wu, J.; Ju, Y.; Sun, X.; Liu, Y.; Shan, B. Blocking the Notch signal transduction pathway promotes tumor growth in osteosarcoma by affecting polarization of TAM to M2 phenotype. Ann. Transl. Med. 2020, 8, 1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, H.; Wang, Y.; Zhan, F.; Quan, Z. Identification of Immune-Related Genes MSR1 and TLR7 in Relation to Macrophage and Type-2 T-Helper Cells in Osteosarcoma Tumor Micro-Environments as Anti-metastasis Signatures. Front. Mol. Biosci. 2020, 7, 576298. [Google Scholar] [CrossRef] [PubMed]
- Dumars, C.; Ngyuen, J.M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef]
- Shao, X.J.; Xiang, S.F.; Chen, Y.Q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.D.; He, Q.J. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Tome, Y.; Kiyuna, T.; Uehara, F.; Bouvet, M.; Tsuchiya, H.; Kanaya, F.; Hoffman, R.M. Imaging the interaction of alphav integrin-GFP in osteosarcoma cells with RFP-expressing host stromal cells and tumor-scaffold collagen in the primary and metastatic tumor microenvironment. J. Cell Biochem. 2019, 120, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Xu, A.; Jin, J.; Zhang, M.; Lou, J.; Qian, C.; Zhu, J.; Wang, Y.; Yang, Z.; Li, X.; et al. MerTK-mediated efferocytosis promotes immune tolerance and tumor progression in osteosarcoma through enhancing M2 polarization and PD-L1 expression. Oncoimmunology 2022, 11, 2024941. [Google Scholar] [CrossRef]
- Li, J.; Zhao, C.; Li, Y.; Wen, J.; Wang, S.; Wang, D.; Dong, H.; Wang, D.; Zhao, Y.; Wang, X.; et al. Osteosarcoma exocytosis of soluble LGALS3BP mediates macrophages toward a tumoricidal phenotype. Cancer Lett. 2022, 528, 1–15. [Google Scholar] [CrossRef]
- Nazon, C.; Pierrevelcin, M.; Willaume, T.; Lhermitte, B.; Weingertner, N.; Marco, A.D.; Bund, L.; Vincent, F.; Bierry, G.; Gomez-Brouchet, A.; et al. Together Intra-Tumor Hypoxia and Macrophagic Immunity Are Driven Worst Outcome in Pediatric High-Grade Osteosarcomas. Cancers 2022, 14, 1482. [Google Scholar] [CrossRef]
- Yang, B.; Su, Z.; Chen, G.; Zeng, Z.; Tan, J.; Wu, G.; Zhu, S.; Lin, L. Identification of prognostic biomarkers associated with metastasis and immune infiltration in osteosarcoma. Oncol. Lett. 2021, 21, 180. [Google Scholar] [CrossRef]
- Le, T.; Su, S.; Kirshtein, A.; Shahriyari, L. Data-Driven Mathematical Model of Osteosarcoma. Cancers 2021, 13, 2367. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Thomson, K.; Pang, P.; Dutour, A.; Mirabello, L.; Acher, F.; Pin, J.P.; Demicco, E.G.; Yan, J.; Teng, M.W.L.; et al. Infiltrating Myeloid Cells Drive Osteosarcoma Progression via GRM4 Regulation of IL23. Cancer Discov. 2019, 9, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef] [PubMed]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Sundara, Y.T.; Kostine, M.; Cleven, A.H.; Bovee, J.V.; Schilham, M.W.; Cleton-Jansen, A.M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: A rationale for T-cell-based immunotherapy. Cancer Immunol. Immunother. 2017, 66, 119–128. [Google Scholar] [CrossRef]
- Han, Q.; Shi, H.; Liu, F. CD163(+) M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016, 34, 101–106. [Google Scholar] [CrossRef]
- Casanova, J.M.; Almeida, J.S.; Reith, J.D.; Sousa, L.M.; Fonseca, R.; Freitas-Tavares, P.; Santos-Rosa, M.; Rodrigues-Santos, P. Tumor-Infiltrating Lymphocytes and Cancer Markers in Osteosarcoma: Influence on Patient Survival. Cancers 2021, 13, 6075. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, Q.; Fan, Y.; Li, J.; Zhang, J.; Wang, W.; Fan, J.; Guo, Y.; Liu, S.; Hao, D.; et al. MYC inhibition reprograms tumor immune microenvironment by recruiting T lymphocytes and activating the CD40/CD40L system in osteosarcoma. Cell Death Discov. 2022, 8, 117. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, Z.; Jing, D.; Huang, X.; Ren, D.; Shao, Z.; Zhang, Z. SGLT2 inhibitor activates the STING/IRF3/IFN-beta pathway and induces immune infiltration in osteosarcoma. Cell Death Dis. 2022, 13, 523. [Google Scholar] [CrossRef]
- Yahiro, K.; Matsumoto, Y.; Yamada, H.; Endo, M.; Setsu, N.; Fujiwara, T.; Nakagawa, M.; Kimura, A.; Shimada, E.; Okada, S.; et al. Activation of TLR4 signaling inhibits progression of osteosarcoma by stimulating CD8-positive cytotoxic lymphocytes. Cancer Immunol. Immunother. 2020, 69, 745–758. [Google Scholar] [CrossRef]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-beta. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Chen, L.; Li, Y.J.; Kong, D.L. PDL1/PD1 axis serves an important role in natural killer cellinduced cytotoxicity in osteosarcoma. Oncol. Rep. 2019, 42, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Li, S.; Li, B.; Sun, L.; Li, H.; Lin, P.; Wang, S.; Teng, W.; Zhou, X.; et al. Decitabine Enhances Vgamma9Vdelta2 T Cell-Mediated Cytotoxic Effects on Osteosarcoma Cells via the NKG2DL-NKG2D Axis. Front. Immunol. 2018, 9, 1239. [Google Scholar] [CrossRef]
- Castella, B.; Melaccio, A.; Foglietta, M.; Riganti, C.; Massaia, M. Vgamma9Vdelta2 T Cells as Strategic Weapons to Improve the Potency of Immune Checkpoint Blockade and Immune Interventions in Human Myeloma. Front. Oncol. 2018, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Yang, C.; Tan, J.; Dou, C.; Chen, Y. Beyond immunosuppressive effects: Dual roles of myeloid-derived suppressor cells in bone-related diseases. Cell Mol. Life Sci. 2021, 78, 7161–7183. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Okamoto, M.; Sasaki, J.; Kuroda, C.; Ishida, H.; Ueda, K.; Ideta, H.; Kamanaka, T.; Sobajima, A.; Takizawa, T.; et al. Anti-PD-1 antibody decreases tumour-infiltrating regulatory T cells. BMC Cancer 2020, 20, 25. [Google Scholar] [CrossRef]
- Chen, K.; Jiao, Y.; Liu, L.; Huang, M.; He, C.; He, W.; Hou, J.; Yang, M.; Luo, X.; Li, C. Communications Between Bone Marrow Macrophages and Bone Cells in Bone Remodeling. Front Cell Dev. Biol. 2020, 8, 598263. [Google Scholar] [CrossRef]
- Su, Y.; Zhou, Y.; Sun, Y.J.; Wang, Y.L.; Yin, J.Y.; Huang, Y.J.; Zhang, J.J.; He, A.N.; Han, K.; Zhang, H.Z.; et al. Macrophage-derived CCL18 promotes osteosarcoma proliferation and migration by upregulating the expression of UCA1. J. Mol. Med. 2019, 97, 49–61. [Google Scholar] [CrossRef]
- Liu, M.X.; Liu, Q.Y.; Liu, Y.; Cheng, Z.M.; Liu, L.; Zhang, L.; Sun, D.H. Interleukin-35 suppresses antitumor activity of circulating CD8(+) T cells in osteosarcoma patients. Connect. Tissue Res. 2019, 60, 367–375. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, J.; Mitra, A.; Cutrera, J.; Zhang, W.; Zhang, Z.; Yan, J.; Xia, X.; Mahadeo, K.M.; Livingston, J.A.; et al. Tumor-targeted IL-12 combined with tumor resection yields a survival-favorable immune profile. J. Immunother. Cancer 2019, 7, 154. [Google Scholar] [CrossRef]
- Chen, L.H.; Liu, J.F.; Lu, Y.; He, X.Y.; Zhang, C.; Zhou, H.H. Complement C1q (C1qA, C1qB, and C1qC) May Be a Potential Prognostic Factor and an Index of Tumor Microenvironment Remodeling in Osteosarcoma. Front. Oncol. 2021, 11, 642144. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Han, S.R.; Lee, S.; Park, S.J.; Kim, J.H.; Yoo, S.M.; Lee, M.S. Activation of the complement system in an osteosarcoma cell line promotes angiogenesis through enhanced production of growth factors. Sci. Rep. 2018, 8, 5415. [Google Scholar] [CrossRef] [PubMed]
- Troyer, R.M.; Ruby, C.E.; Goodall, C.P.; Yang, L.; Maier, C.S.; Albarqi, H.A.; Brady, J.V.; Bathke, K.; Taratula, O.; Mourich, D.; et al. Exosomes from Osteosarcoma and normal osteoblast differ in proteomic cargo and immunomodulatory effects on T cells. Exp. Cell Res. 2017, 358, 369–376. [Google Scholar] [CrossRef]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology 2020, 9, 1747677. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Wang, L.; Wu, C.; Huang, L.; Ruan, Y.; Xue, W. Tumor-derived Exosomes Induced M2 Macrophage Polarization and Promoted the Metastasis of Osteosarcoma Cells Through Tim-3. Arch. Med. Res. 2021, 52, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Yati, S.; Silathapanasakul, A.; Thakaeng, C.; Chanasakulniyom, M.; Songtawee, N.; Porntadavity, S.; Pothacharoen, P.; Pruksakorn, D.; Kongtawelert, P.; Yenchitsomanus, P.T.; et al. Extracellular Vesicle-Mediated IL-1 Signaling in Response to Doxorubicin Activates PD-L1 Expression in Osteosarcoma Models. Cells 2022, 11, 1042. [Google Scholar] [CrossRef] [PubMed]
- Brohl, A.S.; Sindiri, S.; Wei, J.S.; Milewski, D.; Chou, H.C.; Song, Y.K.; Wen, X.; Kumar, J.; Reardon, H.V.; Mudunuri, U.S.; et al. Immuno-transcriptomic profiling of extracranial pediatric solid malignancies. Cell Rep. 2021, 37, 110047. [Google Scholar] [CrossRef]
- Guo, M.; Hartlova, A.; Gierlinski, M.; Prescott, A.; Castellvi, J.; Losa, J.H.; Petersen, S.K.; Wenzel, U.A.; Dill, B.D.; Emmerich, C.H.; et al. Triggering MSR1 promotes JNK-mediated inflammation in IL-4-activated macrophages. EMBO J. 2019, 38, e100299. [Google Scholar] [CrossRef]
- Peng, J.; Tsang, J.Y.; Li, D.; Niu, N.; Ho, D.H.; Lau, K.F.; Lui, V.C.; Lamb, J.R.; Chen, Y.; Tam, P.K. Inhibition of TGF-beta signaling in combination with TLR7 ligation re-programs a tumoricidal phenotype in tumor-associated macrophages. Cancer Lett. 2013, 331, 239–249. [Google Scholar] [CrossRef]
- Liang, T.; Chen, J.; Xu, G.; Zhang, Z.; Xue, J.; Zeng, H.; Jiang, J.; Chen, T.; Qin, Z.; Li, H.; et al. TYROBP, TLR4 and ITGAM regulated macrophages polarization and immune checkpoints expression in osteosarcoma. Sci. Rep. 2021, 11, 19315. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Xu, W.; Wang, X.; Ke, D.; Lin, J.; Lin, W.; Bai, X. Classification of Osteosarcoma Based on Immunogenomic Profiling. Front Cell Dev. Biol. 2021, 9, 696878. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, H.; Chen, Y.; Chen, H.; Yuan, W.; Wang, X. The Heterogeneity of Infiltrating Macrophages in Metastatic Osteosarcoma and Its Correlation with Immunotherapy. J. Oncol. 2021, 2021, 4836292. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; He, G.; Liu, Z.; Wang, J.; Zhang, Z.; Bao, Q.; Wen, J.; Jin, Z.; Zhang, W. Exploration and Validation of a Novel Inflammatory Response-Associated Gene Signature to Predict Osteosarcoma Prognosis and Immune Infiltration. J. Inflamm. Res. 2021, 14, 6719–6734. [Google Scholar] [CrossRef] [PubMed]
- Marchais, A.; Marques da Costa, M.E.; Job, B.; Abbas, R.; Drubay, D.; Piperno-Neumann, S.; Fromigue, O.; Gomez-Brouchet, A.; Redini, F.; Droit, R.; et al. Immune Infiltrate and Tumor Microenvironment Transcriptional Programs Stratify Pediatric Osteosarcoma into Prognostic Groups at Diagnosis. Cancer Res. 2022, 82, 974–985. [Google Scholar] [CrossRef]
- Cai, D.; Ma, X.; Guo, H.; Zhang, H.; Bian, A.; Yu, H.; Cheng, W. Prognostic value of p16, p53, and pcna in sarcoma and an evaluation of immune infiltration. J. Orthop. Surg. Res. 2022, 17, 305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, W.; Zhou, Q.; Chen, C.; Yuan, W.; Liu, J.; Li, X.; Sun, Z. Roles of circRNAs in the tumour microenvironment. Mol. Cancer 2020, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Kousar, K.; Ahmad, T.; Abduh, M.S.; Kanwal, B.; Shah, S.S.; Naseer, F.; Anjum, S. miRNAs in Regulation of Tumor Microenvironment, Chemotherapy Resistance, Immunotherapy Modulation and miRNA Therapeutics in Cancer. Int. J. Mol. Sci. 2022, 23, 13822. [Google Scholar] [CrossRef]
- Chen, Z.; Li, L.; Li, Z.; Wang, X.; Han, M.; Gao, Z.; Wang, M.; Hu, G.; Xie, X.; Du, H.; et al. Identification of key serum biomarkers for the diagnosis and metastatic prediction of osteosarcoma by analysis of immune cell infiltration. Cancer Cell Int. 2022, 22, 78. [Google Scholar] [CrossRef]
- Thakur, M.D.; Franz, C.J.; Brennan, L.; Brouwer-Visser, J.; Tam, R.; Korski, K.; Koeppen, H.; Ziai, J.; Babitzki, G.; Ranchere-Vince, D.; et al. Immune contexture of paediatric cancers. Eur. J. Cancer 2022, 170, 179–193. [Google Scholar] [CrossRef]
- Luo, D.; Ren, H.; Zhang, W.; Xian, H.; Lian, K.; Liu, H. Clinicopathological and prognostic value of hypoxia-inducible factor-1alpha in patients with bone tumor: A systematic review and meta-analysis. J. Orthop. Surg. Res. 2019, 14, 56. [Google Scholar] [CrossRef]
- Pierrevelcin, M.; Fuchs, Q.; Lhermitte, B.; Messe, M.; Guerin, E.; Weingertner, N.; Martin, S.; Lelong-Rebel, I.; Nazon, C.; Dontenwill, M.; et al. Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability. Cells 2020, 9, 1998. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Salaroglio, I.C.; Perez-Ruiz, E.; Sarmento-Ribeiro, A.B.; Saponara, S.; De Las Rivas, J.; Riganti, C. Hypoxia as a driver of resistance to immunotherapy. Drug Resist. Updat. 2021, 59, 100787. [Google Scholar] [CrossRef]
- Fu, Y.; Bao, Q.; Liu, Z.; He, G.; Wen, J.; Liu, Q.; Xu, Y.; Jin, Z.; Zhang, W. Development and Validation of a Hypoxia-Associated Prognostic Signature Related to Osteosarcoma Metastasis and Immune Infiltration. Front. Cell Dev. Biol. 2021, 9, 633607. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Tanaka, K.; Itonaga, I.; Iwasaki, T.; Miyazaki, M.; Ikeda, S.; Tsumura, H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol. Lett. 2016, 11, 2169–2175. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Jin, T.; Wu, H.; Wang, Y.; Peng, H. Capsaicin induces immunogenic cell death in human osteosarcoma cells. Exp. Ther. Med. 2016, 12, 765–770. [Google Scholar] [CrossRef]
- Buondonno, I.; Gazzano, E.; Jean, S.R.; Audrito, V.; Kopecka, J.; Fanelli, M.; Salaroglio, I.C.; Costamagna, C.; Roato, I.; Mungo, E.; et al. Mitochondria-Targeted Doxorubicin: A New Therapeutic Strategy against Doxorubicin-Resistant Osteosarcoma. Mol Cancer Ther. 2016, 15, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Campia, I.; Brusa, D.; Doublier, S.; Matera, L.; Ghigo, D.; Bosia, A.; Riganti, C. Nitric oxide and P-glycoprotein modulate the phagocytosis of colon cancer cells. J. Cell Mol. Med. 2011, 15, 1492–1504. [Google Scholar] [CrossRef]
- Buondonno, I.; Gazzano, E.; Tavanti, E.; Chegaev, K.; Kopecka, J.; Fanelli, M.; Rolando, B.; Fruttero, R.; Gasco, A.; Hattinger, C.; et al. Endoplasmic reticulum-targeting doxorubicin: A new tool effective against doxorubicin-resistant osteosarcoma. Cell Mol. Life Sci. 2019, 76, 609–625. [Google Scholar] [CrossRef]
- Gazzano, E.; Buondonno, I.; Marengo, A.; Rolando, B.; Chegaev, K.; Kopecka, J.; Saponara, S.; Sorge, M.; Hattinger, C.M.; Gasco, A.; et al. Hyaluronated liposomes containing H2S-releasing doxorubicin are effective against P-glycoprotein-positive/doxorubicin-resistant osteosarcoma cells and xenografts. Cancer Lett. 2019, 456, 29–39. [Google Scholar] [CrossRef]
- He, L.; Yang, H.; Huang, J. The tumor immune microenvironment and immune-related signature predict the chemotherapy response in patients with osteosarcoma. BMC Cancer 2021, 21, 581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Cheng, D.; Dang, J.; Mi, Z.; Shi, Y.; Wang, L.; Fan, H. Unfolded Protein Response-Related Signature Associates With the Immune Microenvironment and Prognostic Prediction in Osteosarcoma. Front Genet. 2022, 13, 911346. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Luo, K.; Liu, Y.; Liao, S.; He, J.; He, M.; Xie, T.; Jiang, X.; Li, B.; Liu, H.; et al. ATG16L1 is a Potential Prognostic Biomarker and Immune Signature for Osteosarcoma: A Study Based on Bulk RNA and Single-Cell RNA-Sequencing. Int. J. Gen. Med. 2022, 15, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Castella, B.; Kopecka, J.; Sciancalepore, P.; Mandili, G.; Foglietta, M.; Mitro, N.; Caruso, D.; Novelli, F.; Riganti, C.; Massaia, M. The ATP-binding cassette transporter A1 regulates phosphoantigen release and Vgamma9Vdelta2 T cell activation by dendritic cells. Nat. Commun. 2017, 8, 15663. [Google Scholar] [CrossRef]
- Belisario, D.C.; Akman, M.; Godel, M.; Campani, V.; Patrizio, M.P.; Scotti, L.; Hattinger, C.M.; De Rosa, G.; Donadelli, M.; Serra, M.; et al. ABCA1/ABCB1 Ratio Determines Chemo- and Immune-Sensitivity in Human Osteosarcoma. Cells 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Beird, H.C.; Andrew Livingston, J.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Sun, C.Y.; Zhang, Z.; Tao, L.; Xu, F.F.; Li, H.Y.; Zhang, H.Y.; Liu, W. T cell exhaustion drives osteosarcoma pathogenesis. Ann. Transl. Med. 2021, 9, 1447. [Google Scholar] [CrossRef] [PubMed]
- Ligon, J.A.; Choi, W.; Cojocaru, G.; Fu, W.; Hsiue, E.H.; Oke, T.F.; Siegel, N.; Fong, M.H.; Ladle, B.; Pratilas, C.A.; et al. Pathways of immune exclusion in metastatic osteosarcoma are associated with inferior patient outcomes. J. Immunother. Cancer. 2021, 9, e001772. [Google Scholar] [CrossRef] [PubMed]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019, 15, 100221. [Google Scholar] [CrossRef]
- Nasarre, P.; Garcia, D.I.; Siegel, J.B.; Bonilla, I.V.; Mukherjee, R.; Hilliard, E.; Chakraborty, P.; Nasarre, C.; Yustein, J.T.; Lang, M.; et al. Overcoming PD-1 Inhibitor Resistance with a Monoclonal Antibody to Secreted Frizzled-Related Protein 2 in Metastatic Osteosarcoma. Cancers 2021, 13, 2696. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Zhang, L.; Xue, L.; Wu, Y.; Wu, Q.; Ren, H.; Song, X. Exosomes loaded with programmed death ligand-1 promote tumor growth by immunosuppression in osteosarcoma. Bioengineered 2021, 12, 9520–9530. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Qiu, C.; Wang, J. MicroRNA-519d-3p antagonizes osteosarcoma resistance against cisplatin by targeting PD-L1. Mol. Carcinog. 2022, 61, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, X.; Wang, H.; Yu, Z.; Zhu, Y.; Gao, Y. Specific inhibition of PI3Kdelta/gamma enhances the efficacy of anti-PD1 against osteosarcoma cancer. J. Bone Oncol. 2019, 16, 100206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Yang, S.; Zhang, L.; Wang, W. Anti-CD40 mAb enhanced efficacy of anti-PD1 against osteosarcoma. J. Bone Oncol. 2019, 17, 100245. [Google Scholar] [CrossRef]
- Gul Mohammad, A.; Li, D.; He, R.; Lei, X.; Mao, L.; Zhang, B.; Zhong, X.; Yin, Z.; Cao, W.; Zhang, W.; et al. Integrated analyses of an RNA binding protein-based signature related to tumor immune microenvironment and candidate drugs in osteosarcoma. Am. J. Transl. Res. 2022, 14, 2501–2526. [Google Scholar] [PubMed]
- Zhong, M.; Gao, R.; Zhao, R.; Huang, Y.; Chen, C.; Li, K.; Yu, X.; Nie, D.; Chen, Z.; Liu, X.; et al. BET bromodomain inhibition rescues PD-1-mediated T-cell exhaustion in acute myeloid leukemia. Cell Death Dis. 2022, 13, 671. [Google Scholar] [CrossRef]
- Starzer, A.M.; Berghoff, A.S.; Hamacher, R.; Tomasich, E.; Feldmann, K.; Hatziioannou, T.; Traint, S.; Lamm, W.; Noebauer-Huhmann, I.M.; Furtner, J.; et al. Tumor DNA methylation profiles correlate with response to anti-PD-1 immune checkpoint inhibitor monotherapy in sarcoma patients. J. Immunother. Cancer 2021, 9, e001458. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Koksal, H.; Muller, E.; Inderberg, E.M.; Bruland, O.; Walchli, S. Treating osteosarcoma with CAR T cells. Scand J. Immunol. 2019, 89, e12741. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Cheung, N.V. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J. Hematol. Oncol. 2020, 13, 172. [Google Scholar] [CrossRef] [PubMed]
- Wiebel, M.; Kailayangiri, S.; Altvater, B.; Meltzer, J.; Grobe, K.; Kupich, S.; Rossig, C. Surface expression of the immunotherapeutic target GD2 in osteosarcoma depends on cell confluency. Cancer Rep. 2021, 4, e1394. [Google Scholar] [CrossRef]
- Talbot, L.J.; Chabot, A.; Funk, A.; Nguyen, P.; Wagner, J.; Ross, A.; Tillman, H.; Davidoff, A.; Gottschalk, S.; DeRenzo, C. A Novel Orthotopic Implantation Technique for Osteosarcoma Produces Spontaneous Metastases and Illustrates Dose-Dependent Efficacy of B7-H3-CAR T Cells. Front. Immunol. 2021, 12, 691741. [Google Scholar] [CrossRef]
- Fernandez, L.; Metais, J.Y.; Escudero, A.; Vela, M.; Valentin, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patino-Garcia, A.; et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017, 23, 5824–5835. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, W.; Zhu, J.; Wang, J.; Xia, K.; Liang, C.; Tao, H. Anti-CD166/4-1BB chimeric antigen receptor T cell therapy for the treatment of osteosarcoma. J. Exp. Clin. Cancer Res. 2019, 38, 168. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Front. Oncol. 2018, 8, 453. [Google Scholar] [CrossRef]
- Zhu, J.; Simayi, N.; Wan, R.; Huang, W. CAR T targets and microenvironmental barriers of osteosarcoma. Cytotherapy 2022, 24, 567–576. [Google Scholar] [CrossRef]
- Yang, Q.; Hu, J.; Jia, Z.; Wang, Q.; Wang, J.; Dao, L.H.; Zhang, W.; Zhang, S.; Xia, X.; Gorlick, R.; et al. Membrane-Anchored and Tumor-Targeted IL12 (attIL12)-PBMC Therapy for Osteosarcoma. Clin. Cancer Res. 2022, 28, 3862–3873. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef]
- Jiang, K.; Li, J.; Zhang, J.; Wang, L.; Zhang, Q.; Ge, J.; Guo, Y.; Wang, B.; Huang, Y.; Yang, T.; et al. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int. Immunopharmacol. 2019, 75, 105818. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Itonaga, I.; Iwasaki, T.; Tsuchiya, H.; Tsumura, H. Anti-TGF-beta antibody combined with dendritic cells produce antitumor effects in osteosarcoma. Clin. Orthop. Relat. Res. 2012, 470, 2288–2294. [Google Scholar] [CrossRef]
- Zhou, Y.; Slone, N.; Chrisikos, T.T.; Kyrysyuk, O.; Babcock, R.L.; Medik, Y.B.; Li, H.S.; Kleinerman, E.S.; Watowich, S.S. Vaccine efficacy against primary and metastatic cancer with in vitro-generated CD103(+) conventional dendritic cells. J. Immunother. Cancer 2020, 8, e000474. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Yasui, T.; Tamari, K.; Minami, K.; Otani, K.; Isohashi, F.; Seo, Y.; Kambe, R.; Koizumi, M.; Ogawa, K. Radiation enhanced the local and distant anti-tumor efficacy in dual immune checkpoint blockade therapy in osteosarcoma. PLoS ONE 2017, 12, e0189697. [Google Scholar] [CrossRef]
- He, X.; Lin, H.; Yuan, L.; Li, B. Combination therapy with L-arginine and alpha-PD-L1 antibody boosts immune response against osteosarcoma in immunocompetent mice. Cancer Biol. Ther. 2017, 18, 94–100. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Pol, J.G.; Lichty, B.D.; Cummings, D.T.; Mossman, K.L. Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol. Res. 2013, 1, 309–319. [Google Scholar] [CrossRef]
- Kopecka, J.; Godel, M.; Dei, S.; Giampietro, R.; Belisario, D.C.; Akman, M.; Contino, M.; Teodori, E.; Riganti, C. Insights into P-Glycoprotein Inhibitors: New Inducers of Immunogenic Cell Death. Cells 2020, 9, 1033. [Google Scholar] [CrossRef]
- Sukkurwala, A.Q.; Adjemian, S.; Senovilla, L.; Michaud, M.; Spaggiari, S.; Vacchelli, E.; Baracco, E.E.; Galluzzi, L.; Zitvogel, L.; Kepp, O.; et al. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology 2014, 3, e28473. [Google Scholar] [CrossRef]
- Yu, Z.; Geng, J.; Zhang, M.; Zhou, Y.; Fan, Q.; Chen, J. Treatment of osteosarcoma with microwave thermal ablation to induce immunogenic cell death. Oncotarget 2014, 5, 6526–6539. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Porto, S.; Lusa, S.; Gazzano, E.; Salzano, G.; Pinzon-Daza, M.L.; Giordano, A.; Desiderio, V.; Ghigo, D.; De Rosa, G.; et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: A combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget 2016, 7, 20753–20772. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, W.; Zhou, X.; Fu, J.; He, C. Tumor cell membrane-camouflaged responsive nanoparticles enable MRI-guided immuno-chemodynamic therapy of orthotopic osteosarcoma. Bioact. Mater. 2022, 17, 221–233. [Google Scholar] [CrossRef]
- Ge, Y.X.; Zhang, T.W.; Zhou, L.; Ding, W.; Liang, H.F.; Hu, Z.C.; Chen, Q.; Dong, J.; Xue, F.F.; Yin, X.F.; et al. Enhancement of anti-PD-1/PD-L1 immunotherapy for osteosarcoma using an intelligent autophagy-controlling metal organic framework. Biomaterials 2022, 282, 121407. [Google Scholar] [CrossRef]
- Jin, J.; Yuan, P.; Yu, W.; Lin, J.; Xu, A.; Xu, X.; Lou, J.; Yu, T.; Qian, C.; Liu, B.; et al. Mitochondria-Targeting Polymer Micelle of Dichloroacetate Induced Pyroptosis to Enhance Osteosarcoma Immunotherapy. ACS Nano 2022, 16, 10327–10340. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, Y.; Tazawa, H.; Demiya, K.; Kure, M.; Kondo, H.; Komatsubara, T.; Sugiu, K.; Hasei, J.; Yoshida, A.; Kunisada, T.; et al. Telomerase-specific oncolytic immunotherapy for promoting efficacy of PD-1 blockade in osteosarcoma. Cancer Immunol. Immunother. 2021, 70, 1405–1417. [Google Scholar] [CrossRef]
- Xiao, H.; Jensen, P.E.; Chen, X. Elimination of Osteosarcoma by Necroptosis with Graphene Oxide-Associated Anti-HER2 Antibodies. Int. J. Mol. Sci. 2019, 20, 4360. [Google Scholar] [CrossRef]
- Fessler, E.; Dijkgraaf, F.E.; De Sousa, E.M.F.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett. 2013, 341, 97–104. [Google Scholar] [CrossRef]
- Susen, R.M.; Bauer, R.; Olesch, C.; Fuhrmann, D.C.; Fink, A.F.; Dehne, N.; Jain, A.; Ebersberger, I.; Schmid, T.; Brune, B. Macrophage HIF-2alpha regulates tumor-suppressive Spint1 in the tumor microenvironment. Mol. Carcinog. 2019, 58, 2127–2138. [Google Scholar] [CrossRef]
- Wei, S.; Lu, J.; Lou, J.; Shi, C.; Mo, S.; Shao, Y.; Ni, J.; Zhang, W.; Cheng, X. Gastric Cancer Tumor Microenvironment Characterization Reveals Stromal-Related Gene Signatures Associated With Macrophage Infiltration. Front. Genet. 2020, 11, 663. [Google Scholar] [CrossRef]
- Punzo, F.; Bellini, G.; Tortora, C.; Pinto, D.D.; Argenziano, M.; Pota, E.; Paola, A.D.; Martino, M.D.; Rossi, F. Mifamurtide and TAM-like macrophages: Effect on proliferation, migration and differentiation of osteosarcoma cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Ren, T.; Huang, Y.; Liang, X.; Wang, W.; Niu, J.; Han, Y.; Guo, W. Development of a prognostic gene signature based on an immunogenomic infiltration analysis of osteosarcoma. J. Cell Mol. Med. 2020, 24, 11230–11242. [Google Scholar] [CrossRef]
- Pahl, J.H.; Kwappenberg, K.M.; Varypataki, E.M.; Santos, S.J.; Kuijjer, M.L.; Mohamed, S.; Wijnen, J.T.; van Tol, M.J.; Cleton-Jansen, A.M.; Egeler, R.M.; et al. Macrophages inhibit human osteosarcoma cell growth after activation with the bacterial cell wall derivative liposomal muramyl tripeptide in combination with interferon-gamma. J. Exp. Clin. Cancer Res. 2014, 33, 27. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Lefebvre, M.L.; Labroquere, K.; Faure, O.; Abastado, J.P. Liposomal muramyl tripeptide phosphatidylethanolamine: Targeting and activating macrophages for adjuvant treatment of osteosarcoma. Curr. Cancer Drug Targets 2006, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Burger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Mori, K.; Corradini, N.; Redini, F.; Heymann, D. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin. Pharmacother. 2011, 12, 285–292. [Google Scholar] [CrossRef]
- Anderson, P.M.; Meyers, P.; Kleinerman, E.; Venkatakrishnan, K.; Hughes, D.P.; Herzog, C.; Huh, W.; Sutphin, R.; Vyas, Y.M.; Shen, V.; et al. Mifamurtide in metastatic and recurrent osteosarcoma: A patient access study with pharmacokinetic, pharmacodynamic, and safety assessments. Pediatr. Blood Cancer 2014, 61, 238–244. [Google Scholar] [CrossRef]
- Jimmy, R.; Stern, C.; Lisy, K.; White, S. Effectiveness of mifamurtide in addition to standard chemotherapy for high-grade osteosarcoma: A systematic review. JBI Database Syst. Rev. Implement Rep. 2017, 15, 2113–2152. [Google Scholar] [CrossRef]
- Kager, L.; Potschger, U.; Bielack, S. Review of mifamurtide in the treatment of patients with osteosarcoma. Ther. Clin. Risk Manag. 2010, 6, 279–286. [Google Scholar] [CrossRef]
- Palmerini, E.; Meazza, C.; Tamburini, A.; Bisogno, G.; Ferraresi, V.; Asaftei, S.D.; Milano, G.M.; Coccoli, L.; Manzitti, C.; Luksch, R.; et al. Phase 2 study for nonmetastatic extremity high-grade osteosarcoma in pediatric and adolescent and young adult patients with a risk-adapted strategy based on ABCB1/P-glycoprotein expression: An Italian Sarcoma Group trial (ISG/OS-2). Cancer 2022, 128, 1958–1966. [Google Scholar] [CrossRef]
- Pakos, E.E.; Ioannidis, J.P. The association of P-glycoprotein with response to chemotherapy and clinical outcome in patients with osteosarcoma. A meta-analysis. Cancer 2003, 98, 581–589. [Google Scholar] [CrossRef]
- Serra, M.; Pasello, M.; Manara, M.C.; Scotlandi, K.; Ferrari, S.; Bertoni, F.; Mercuri, M.; Alvegard, T.A.; Picci, P.; Bacci, G.; et al. May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int. J. Oncol. 2006, 29, 1459–1468. [Google Scholar] [CrossRef]
- Serra, M.; Scotlandi, K.; Reverter-Branchat, G.; Ferrari, S.; Manara, M.C.; Benini, S.; Incaprera, M.; Bertoni, F.; Mercuri, M.; Briccoli, A.; et al. Value of P-glycoprotein and clinicopathologic factors as the basis for new treatment strategies in high-grade osteosarcoma of the extremities. J. Clin. Oncol. 2003, 21, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.J.; Dutton, P.; Bruland, O.; Gelderblom, H.; Faleti, A.; Buhnemann, C.; van Maldegem, A.; Johnson, H.; Poulton, L.; Love, S.; et al. Outcomes from a mechanistic biomarker multi-arm and randomised study of liposomal MTP-PE (Mifamurtide) in metastatic and/or recurrent osteosarcoma (EuroSarc-Memos trial). BMC Cancer 2022, 22, 629. [Google Scholar] [CrossRef] [PubMed]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.F.; Pan, X.H.; Zhang, S.J.; Zhao, C.; Qiu, B.S.; Gu, H.F.; Hong, J.F.; Cao, L.; Chen, Y.; Xia, B.; et al. CD47 blockade inhibits tumor progression human osteosarcoma in xenograft models. Oncotarget 2015, 6, 23662–23670. [Google Scholar] [CrossRef]
- Garcia-Aranda, M.; Redondo, M. Targeting Protein Kinases to Enhance the Response to anti-PD-1/PD-L1 Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2296. [Google Scholar] [CrossRef]
- Perez-Ruiz, E.; Melero, I.; Kopecka, J.; Sarmento-Ribeiro, A.B.; Garcia-Aranda, M.; De Las Rivas, J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: Targets, biomarkers, and remedies. Drug Resist. Updat. 2020, 53, 100718. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Boye, K.; Longhi, A.; Guren, T.; Lorenz, S.; Naess, S.; Pierini, M.; Taksdal, I.; Lobmaier, I.; Cesari, M.; Paioli, A.; et al. Pembrolizumab in advanced osteosarcoma: Results of a single-arm, open-label, phase 2 trial. Cancer Immunol. Immunother. 2021, 70, 2617–2624. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Le Cesne, A.; Marec-Berard, P.; Blay, J.Y.; Gaspar, N.; Bertucci, F.; Penel, N.; Bompas, E.; Cousin, S.; Toulmonde, M.; Bessede, A.; et al. Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: Results from the PEMBROSARC study. Eur. J. Cancer 2019, 119, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer 2020, 8, e000798. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Solis Soto, L.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef]
- Poon, V.I.; Roth, M.; Piperdi, S.; Geller, D.; Gill, J.; Rudzinski, E.R.; Hawkins, D.S.; Gorlick, R. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clin. Sarcoma Res. 2015, 5, 4. [Google Scholar] [CrossRef]
- Roth, M.; Linkowski, M.; Tarim, J.; Piperdi, S.; Sowers, R.; Geller, D.; Gill, J.; Gorlick, R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer 2014, 120, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, C.K.; Appel, N.; Labban, N.; Lussier, D.M.; Blattman, J.N.; Hingorani, P. Progress and opportunities for immune therapeutics in osteosarcoma. Immunotherapy 2016, 8, 1233–1244. [Google Scholar] [CrossRef]
- Wedekind, M.F.; Wagner, L.M.; Cripe, T.P. Immunotherapy for osteosarcoma: Where do we go from here? Pediatr. Blood Cancer 2018, 65, e27227. [Google Scholar] [CrossRef]
- Hingorani, P.; Krailo, M.; Buxton, A.; Hutson, P.; Sondel, P.M.; Diccianni, M.; Yu, A.; Morris, C.D.; Womer, R.B.; Crompton, B.; et al. Phase 2 study of anti-disialoganglioside antibody, dinutuximab, in combination with GM-CSF in patients with recurrent osteosarcoma: A report from the Children’s Oncology Group. Eur. J. Cancer 2022, 172, 264–275. [Google Scholar] [CrossRef]
- Moriarity, B.S.; Otto, G.M.; Rahrmann, E.P.; Rathe, S.K.; Wolf, N.K.; Weg, M.T.; Manlove, L.A.; LaRue, R.S.; Temiz, N.A.; Molyneux, S.D.; et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat. Genet. 2015, 47, 615–624. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.G.; Pellicciotta, I.; Hirbe, A.C.; Pradhan, J.; Chen, A.; Rabadan, R.; Schwartz, G.K. A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors. Clin. Cancer Res. 2021, 27, 5519–5527. [Google Scholar] [CrossRef]
- Sharma, M.D.; Pacholczyk, R.; Shi, H.; Berrong, Z.J.; Zakharia, Y.; Greco, A.; Chang, C.S.; Eathiraj, S.; Kennedy, E.; Cash, T.; et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity 2021, 54, 2354–2371.e2358. [Google Scholar] [CrossRef] [PubMed]
- Thakar, M.S.; Browning, M.; Hari, P.; Charlson, J.A.; Margolis, D.A.; Logan, B.; Schloemer, N.; Kelly, M.E.; Newman, A.; Johnson, B.; et al. Phase II trial using haploidentical hematopoietic cell transplantation (HCT) followed by donor natural killer (NK) cell infusion and sirolimus maintenance for patients with high-risk solid tumors. J. Clin. Oncol. 2020, 38, e23551. [Google Scholar] [CrossRef]
- Amaria, R.N.; Bernatchez, C.; Forget, M.-A.; Haymaker, C.L.; Conley, A.P.; Livingston, J.A.; Varadhachary, G.R.; Javle, M.M.; Maitra, A.; Tzeng, C.-W.D.; et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with sarcomas, ovarian, and pancreatic cancers. J. Clin. Oncol. 2019, 37, TPS2650. [Google Scholar] [CrossRef]
- Dhir, A.; Koru-Sengul, T.; Grosso, J.; D’Amato, G.Z.; Trucco, M.M.; Rosenberg, A.; Gilboa, E.; Goldberg, J.M.; Trent, J.C.; Wilky, B.A. Phase 1 trial of autologous dendritic cell vaccination with imiquimod immunomodulation in children and adults with refractory sarcoma. J. Clin. Oncol. 2021, 39, 11542. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hattinger, C.M.; Salaroglio, I.C.; Fantoni, L.; Godel, M.; Casotti, C.; Kopecka, J.; Scotlandi, K.; Ibrahim, T.; Riganti, C.; Serra, M. Strategies to Overcome Resistance to Immune-Based Therapies in Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 799. https://doi.org/10.3390/ijms24010799
Hattinger CM, Salaroglio IC, Fantoni L, Godel M, Casotti C, Kopecka J, Scotlandi K, Ibrahim T, Riganti C, Serra M. Strategies to Overcome Resistance to Immune-Based Therapies in Osteosarcoma. International Journal of Molecular Sciences. 2023; 24(1):799. https://doi.org/10.3390/ijms24010799
Chicago/Turabian StyleHattinger, Claudia Maria, Iris Chiara Salaroglio, Leonardo Fantoni, Martina Godel, Chiara Casotti, Joanna Kopecka, Katia Scotlandi, Toni Ibrahim, Chiara Riganti, and Massimo Serra. 2023. "Strategies to Overcome Resistance to Immune-Based Therapies in Osteosarcoma" International Journal of Molecular Sciences 24, no. 1: 799. https://doi.org/10.3390/ijms24010799
APA StyleHattinger, C. M., Salaroglio, I. C., Fantoni, L., Godel, M., Casotti, C., Kopecka, J., Scotlandi, K., Ibrahim, T., Riganti, C., & Serra, M. (2023). Strategies to Overcome Resistance to Immune-Based Therapies in Osteosarcoma. International Journal of Molecular Sciences, 24(1), 799. https://doi.org/10.3390/ijms24010799