
Target-Specific Nanoparticle Polyplex Down-Regulates Mutant Kras to Prevent Pancreatic Carcinogenesis and Halt Tumor Progression

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
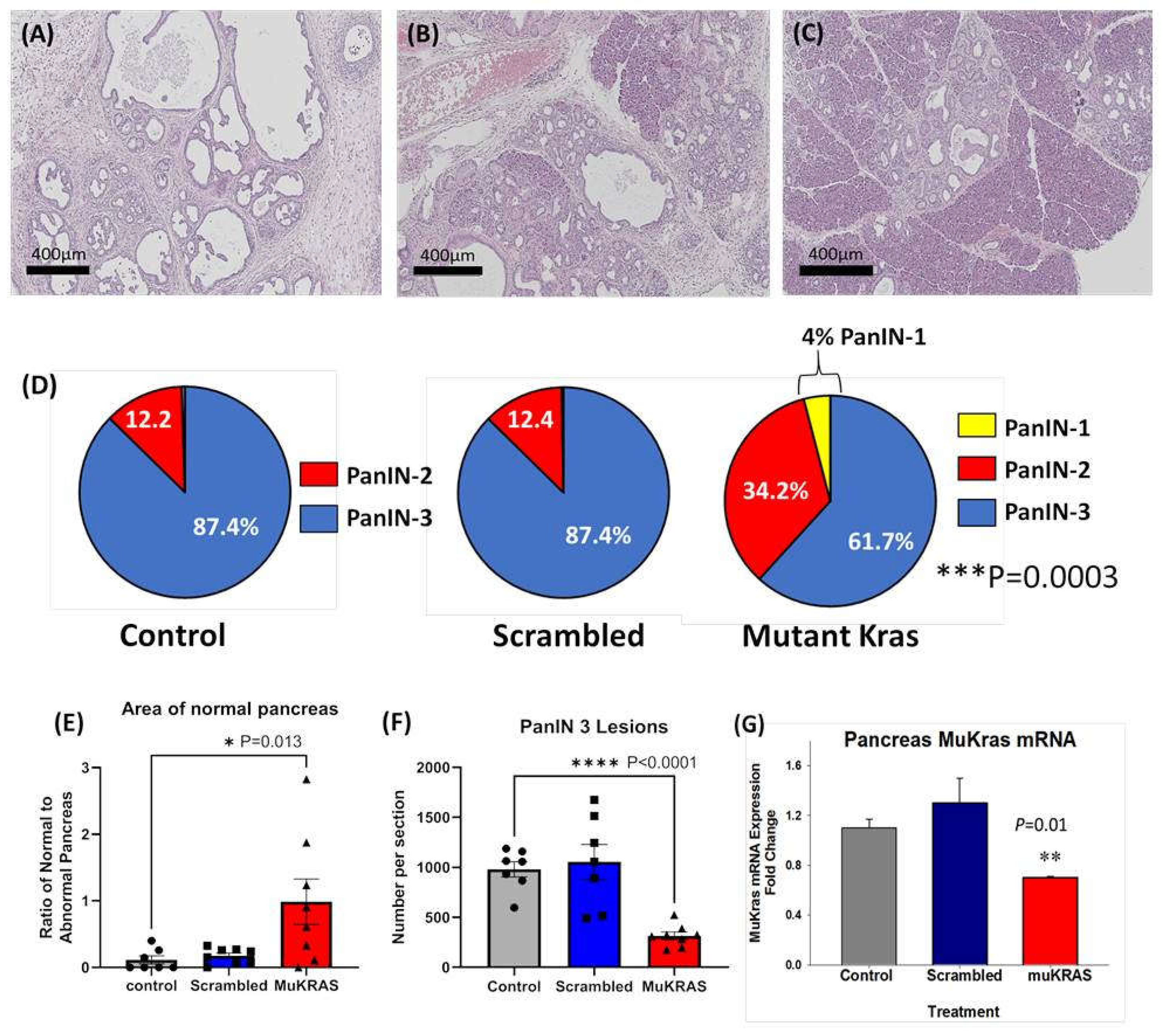
2.1. Targeted NPs with siRNA to Mutant Kras Prevent PanIN Progression
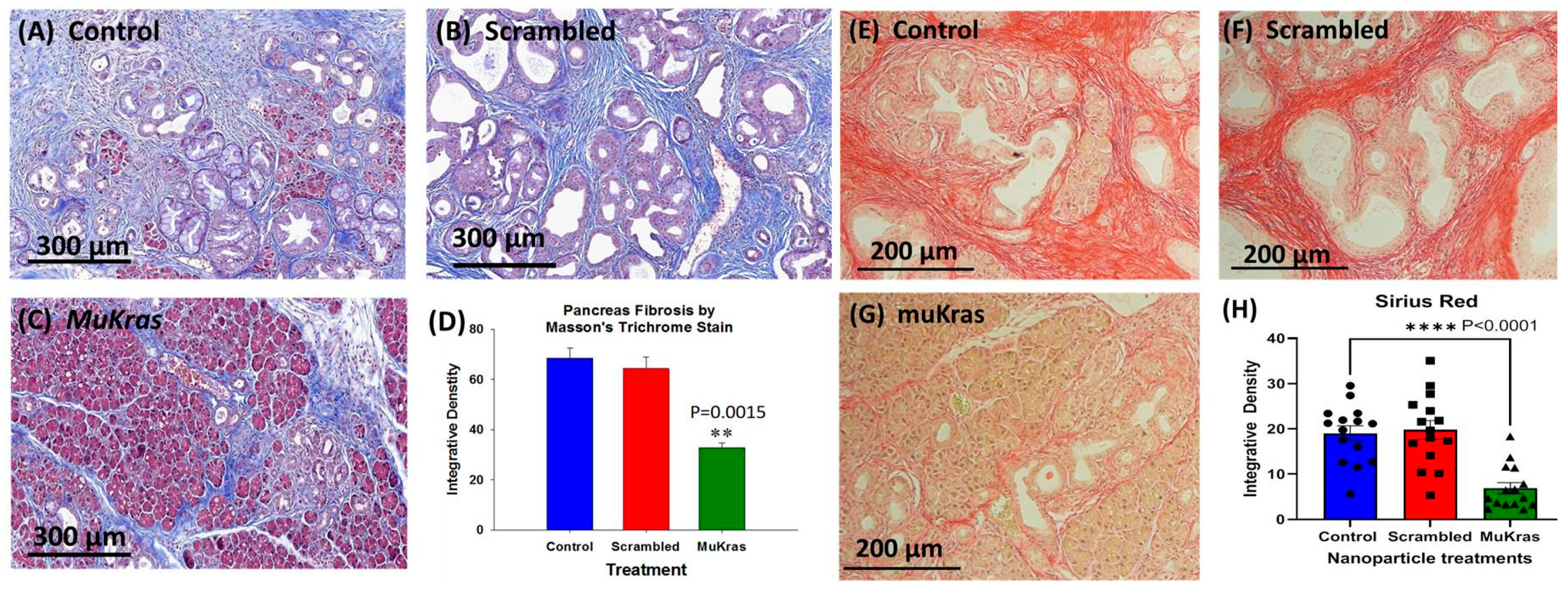
2.2. Targeted NPs Loaded with Mutant Kras siRNA Decrease Fibrosis in the Pancreas Extracellular Matrix
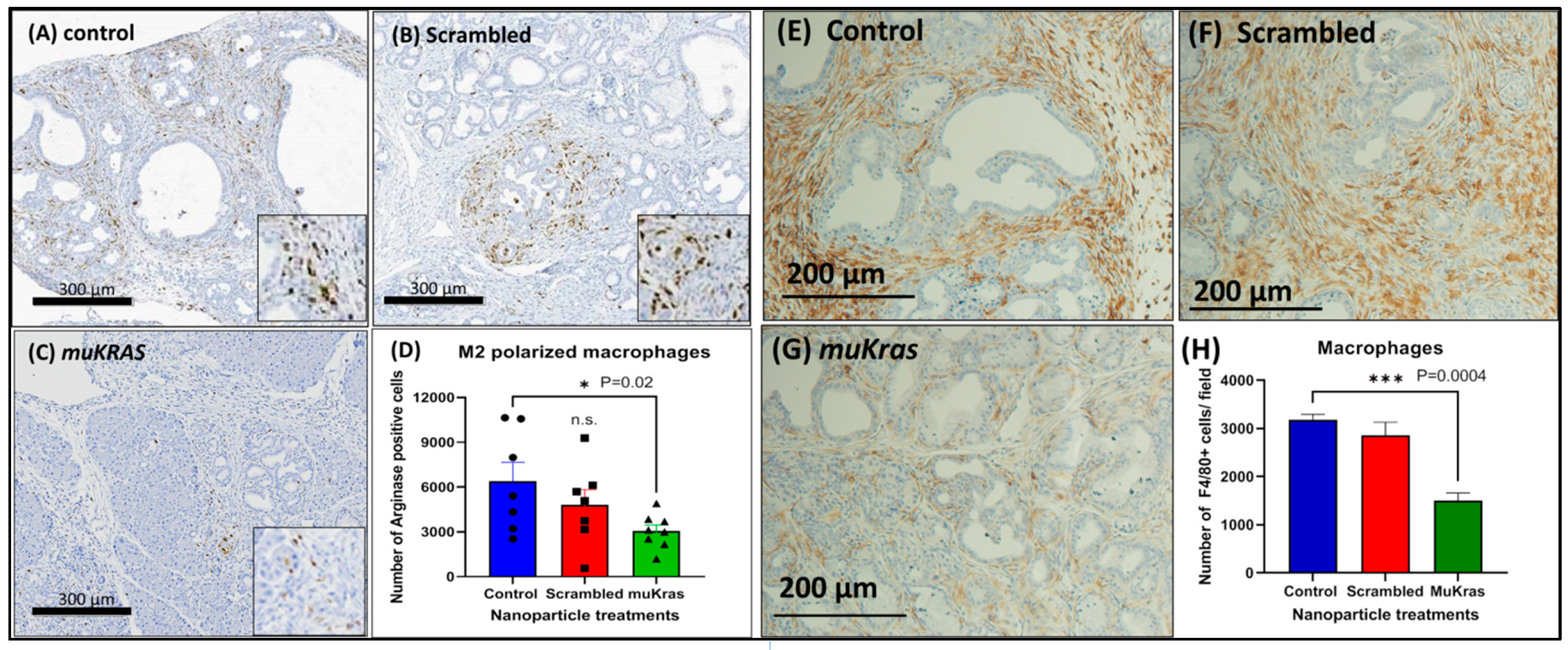
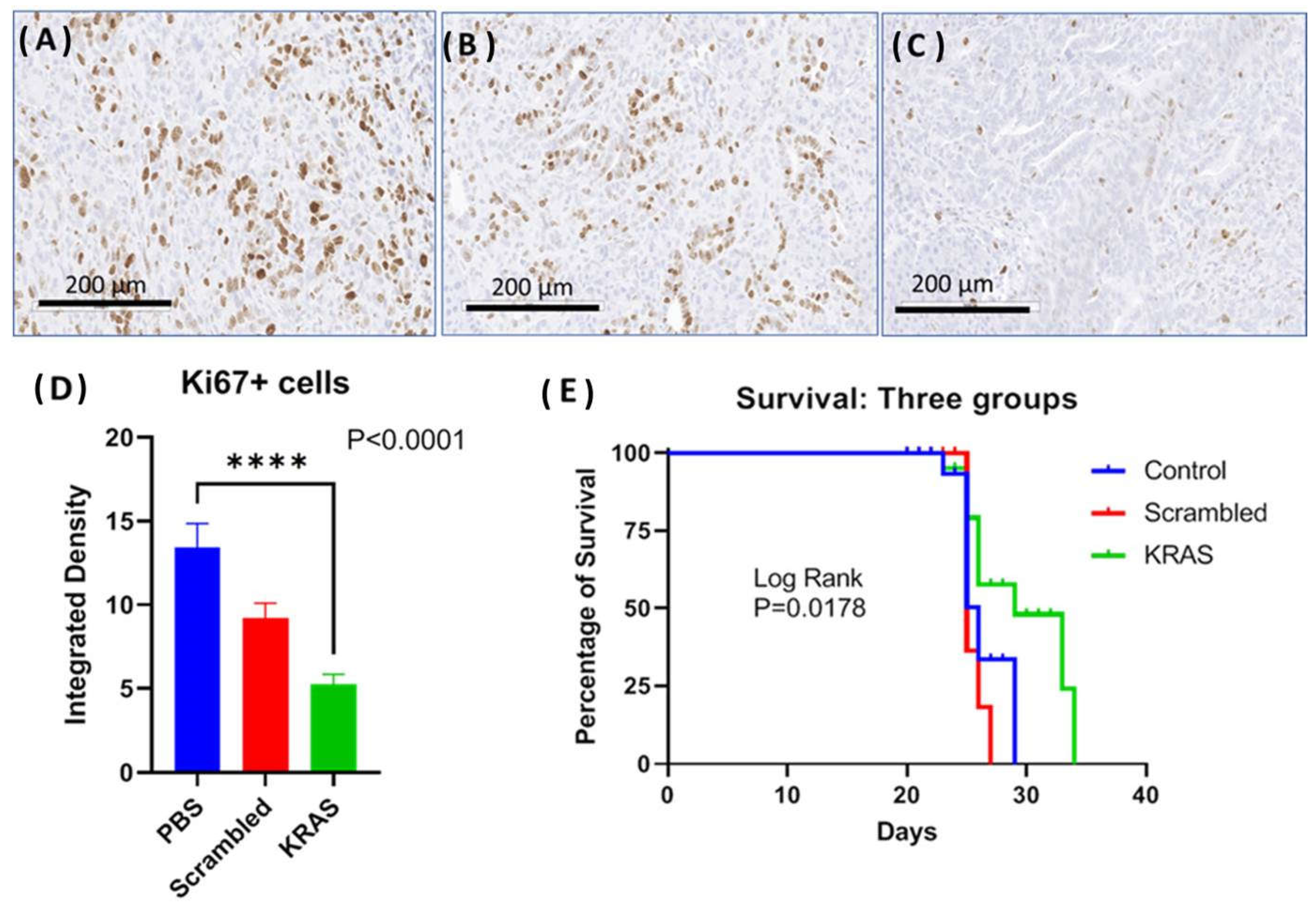
2.3. Targeted NPs Loaded with muKras siRNA Alter Immune Cell Signature by Decreasing M2-Polarized Macrophages
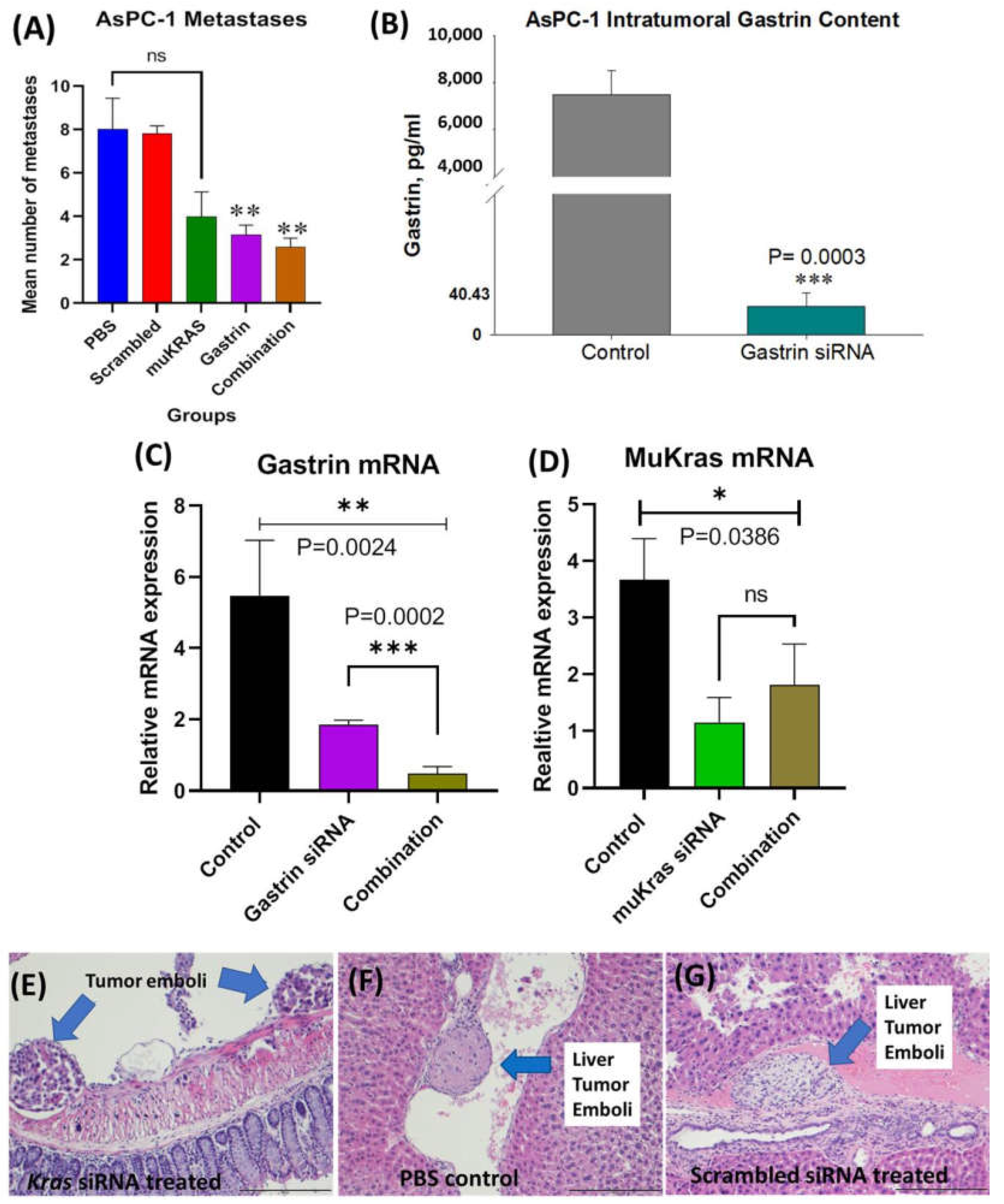
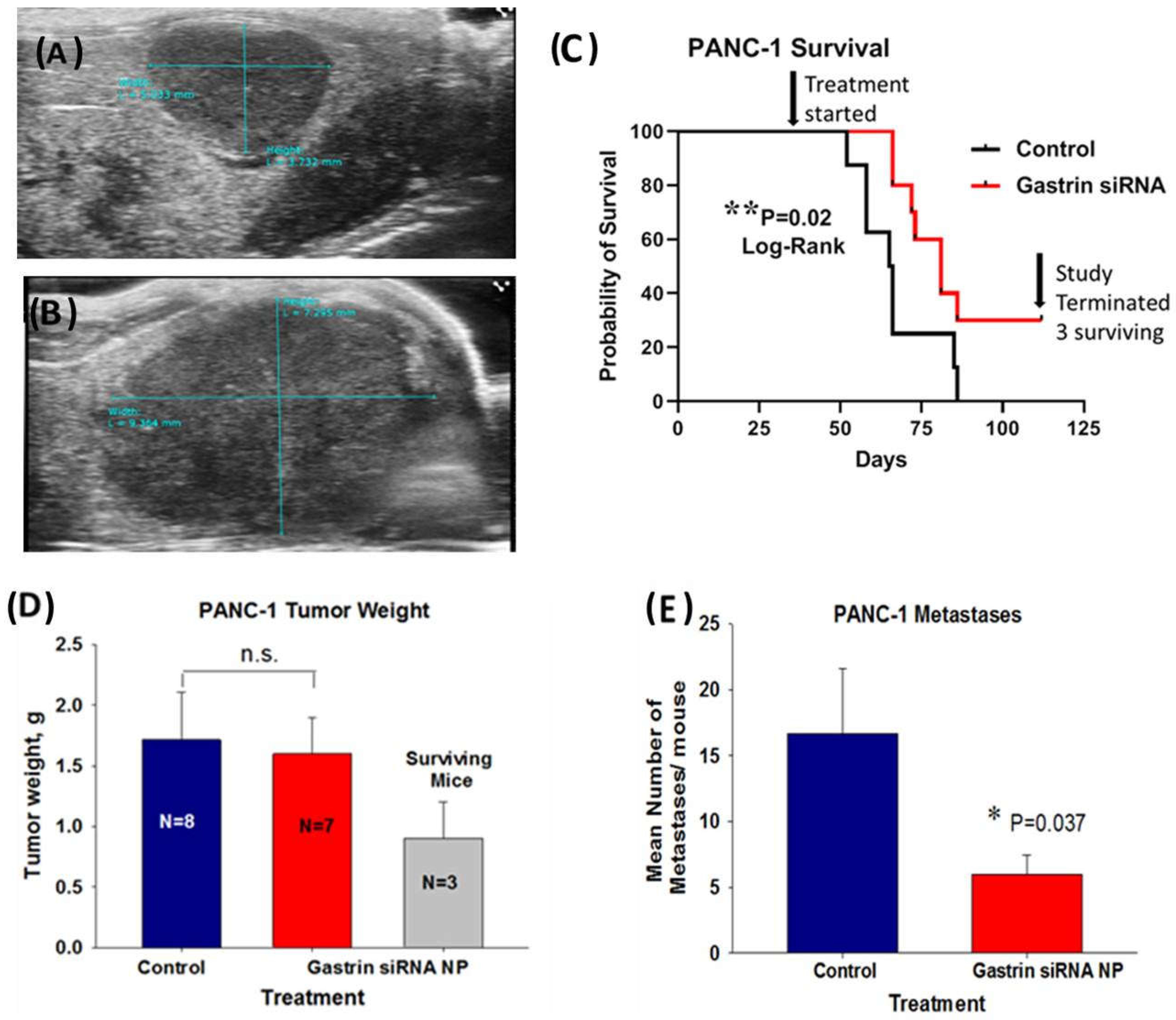
2.4. CCK-BR Targeted NP Improves Survival and Decreases Metastases of Orthotopic Human Pancreatic Cancer Tumors in Mice
2.5. Targeted NP Therapy with Kras siRNA Improves Survival in an Immune Competent Mouse
2.6. CCK-BR Targeted NPs Exhibit Broad Safety Profile and Lack off Target Toxicity
3. Discussion
4. Materials and Methods
4.1. Characterization of Cell Lines
4.2. Nanoparticle Polyplex (NP) Formulation and Characterization
4.3. In Vivo Animal Models
4.4. Evaluation of the Effectiveness of Targeted NPs with siRNA to MuKras to Prevent PanIN Progression
4.5. Orthotopic Pancreatic Cancer Experiments
4.6. Confirmation of Gastrin Peptide Down-Regulation by ELISA
4.7. Confirmation That the siRNA Was Delivered and Down-Regulated the Gene by qRT-PCR
4.8. Immunohistochemistry Evaluation of Proliferation Index and M2-Polarized Macrophages
4.9. Safety and Toxicity Assessment
4.10. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Ayres, P.M.; Chio, I.I.C. Metastasis in Pancreatic Ductal Adenocarcinoma: Current Standing and Methodologies. Genes 2019, 11, 6. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef]
- Zeng, S.; Pottler, M.; Lan, B.; Grutzmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; di Magliano, M.P.; Simeone, D.M. Deciphering the role of stroma in pancreatic cancer. Curr. Opin. Gastroenterol. 2013, 29, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Bayat, M.R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben, A.M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Ko, A.; Reni, M.; Riess, H.; Pelzer, U.; O’Reilly, E.M.; Winter, J.; Tempero, M.A. Tempero A RMHReal: APACT: Phase III, multicenter, international, open-label, randomized trial of adjuvant nab-paclitaxel plus gemcitabine (nab-P/G) vs gemcitabine (G) for surgically resected pancreatic adenocarcinoma. PANCREAS 2019, 48, 4000. [Google Scholar]
- Li, X.; Huang, D.B.; Zhang, Q.; Guo, C.X.; Fu, Q.H.; Zhang, X.C.; Tang, T.Y.; Su, W.; Chen, Y.W.; Chen, W.; et al. The efficacy and toxicity of chemotherapy in the elderly with advanced pancreatic cancer. Pancreatology 2020, 20, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.; Molpeceres, J.; Rijo, P.; Reis, C.P. Pancreatic Cancer Therapy Review: From Classic Therapeutic Agents to Modern Nanotechnologies. Curr. Drug Metab. 2017, 18, 346–359. [Google Scholar] [CrossRef]
- Zhu, L.; Staley, C.; Kooby, D.; El-Rays, B.; Mao, H.; Yang, L. Current status of biomarker and targeted nanoparticle development: The precision oncology approach for pancreatic cancer therapy. Cancer Lett. 2017, 388, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.; Bouvet, M. Nanoparticle albumin-bound-paclitaxel: A limited improvement under the current therapeutic paradigm of pancreatic cancer. Expert Opin. Pharmacother. 2015, 16, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ramanathan, R.K.; Borad, M.J.; Laheru, D.A.; Smith, L.S.; Wood, T.E.; Korn, R.L.; Desai, N.; Trieu, V.; Iglesias, J.L.; et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: A phase I/II trial. J. Clin. Oncol. 2011, 29, 4548–4554. [Google Scholar] [CrossRef]
- Min, S.Y.; Byeon, H.J.; Lee, C.; Seo, J.; Lee, E.S.; Shin, B.S.; Choi, H.G.; Lee, K.C.; Youn, Y.S. Facile one-pot formulation of TRAIL-embedded paclitaxel-bound albumin nanoparticles for the treatment of pancreatic cancer. Int. J. Pharm 2015, 494, 506–515. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Piktel, E.; Niemirowicz, K.; Watek, M.; Wollny, T.; Deptula, P.; Bucki, R. Recent insights in nanotechnology-based drugs and formulations designed for effective anti-cancer therapy. J. Nanobiotechnology 2016, 14, 39. [Google Scholar] [CrossRef]
- Burks, J.; Nadella, S.; Mahmud, A.; Mankongpaisarnrung, C.; Wang, J.; Hahm, J.I.; Tucker, R.D.; Shivapurkar, N.; Stern, S.T.; Smith, J.P. Cholecystokinin Receptor-Targeted Polyplex Nanoparticle Inhibits Growth and Metastasis of Pancreatic Cancer. Cell Mol. Gastroenterol Hepatol. 2018, 6, 17–32. [Google Scholar] [CrossRef]
- Smith, J.P.; Rickabaugh, C.A.; McLaughlin, P.J.; Zagon, I.S. Cholecystokinin receptors and PANC-1 human pancreatic cancer cells. Am. J. Physiol. 1993, 265, G149–G155. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Liu, G.; Soundararajan, V.; McLaughlin, P.J.; Zagon, I.S. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am. J. Physiol. 1994, 266, R277–R283. [Google Scholar] [CrossRef] [PubMed]
- Matters, G.L.; Harms, J.F.; McGovern, C.O.; Jayakumar, C.; Crepin, K.; Smith, Z.P.; Nelson, M.C.; Stock, H.; Fenn, C.W.; Kaiser, J.; et al. Growth of human pancreatic cancer is inhibited by down-regulation of gastrin gene expression. Pancreas 2009, 38, e151–e161. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Shih, A.; Wu, Y.; McLaughlin, P.J.; Zagon, I.S. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am. J. Physiol. 1996, 270, R1078–R1084. [Google Scholar] [CrossRef]
- Smith, J.P.; Cooper, T.K.; McGovern, C.O.; Gilius, E.L.; Zhong, Q.; Liao, J.; Molinolo, A.A.; Gutkind, J.S.; Matters, G.L. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas 2014, 43, 1050–1059. [Google Scholar] [CrossRef]
- Smith, J.P.; Cao, H.; Edmondson, E.F.; Dasa, S.S.K.; Stern, S.T. Cholecystokinin-B Receptor-Targeted Nanoparticle for Imaging and Detection of Precancerous Lesions in the Pancreas. Biomolecules 2021, 11, 1766. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2209–2215. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Matters, G.L.; McGovern, C.; Harms, J.F.; Markovic, K.; Anson, K.; Jayakumar, C.; Martenis, M.; Awad, C.; Smith, J.P. Role of endogenous cholecystokinin on growth of human pancreatic cancer. Int. J. Oncol. 2011, 38, 593–601. [Google Scholar] [PubMed]
- Rejiba, S.; Wack, S.; Aprahamian, M.; Hajri, A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci. 2007, 98, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Pecot, C.V.; Wu, S.Y.; Bellister, S.; Filant, J.; Rupaimoole, R.; Hisamatsu, T.; Bhattacharya, R.; Maharaj, A.; Azam, S.; Rodriguez-Aguayo, C.; et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther. 2014, 13, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004, 5, 375–387. [Google Scholar] [CrossRef]
- Hruban, R.H.; Maitra, A.; Goggins, M. Update on pancreatic intraepithelial neoplasia. Int. J. Clin. Exp. Pathol. 2008, 1, 306–316. [Google Scholar]
- Brune, K.; Abe, T.; Canto, M.; O’Malley, L.; Klein, A.P.; Maitra, A.; Volkan, A.N.; Fishman, E.K.; Cameron, J.L.; Yeo, C.J.; et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am. J. Surg. Pathol. 2006, 30, 1067–1076. [Google Scholar]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Blaker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef]
- Nadella, S.; Burks, J.; Al-Sabban, A.; Inyang, G.; Wang, J.; Tucker, R.D.; Zamanis, M.E.; Bukowski, W.; Shivapurkar, N.; Smith, J.P. Dietary fat stimulates pancreatic cancer growth and promotes fibrosis of the tumor microenvironment through the cholecystokinin receptor. Am. J. Physiol Gastrointest Liver Physiol. 2018, 315, G699–G712. [Google Scholar] [CrossRef]
- Malchiodi, Z.X.; Cao, H.; Gay, M.D.; Safronenka, A.; Bansal, S.; Tucker, R.D.; Weinberg, B.A.; Cheema, A.; Shivapurkar, N.; Smith, J.P. Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment. Cancers 2021, 13, 4949. [Google Scholar] [CrossRef]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.V.; Mielgo, A. Macrophages and Fibroblasts, Key Players in Cancer Chemoresistance. Front Cell Dev. Biol. 2018, 6, 131. [Google Scholar] [CrossRef]
- Nadella, S.; Burks, J.; Huber, M.; Wang, J.; Cao, H.; Kallakury, B.; Tucker, R.D.; Boca, S.M.; Jermusyck, A.; Collins, I.; et al. Endogenous Gastrin Collaborates With Mutant KRAS in Pancreatic Carcinogenesis. Pancreas 2019, 48, 894–903. [Google Scholar] [CrossRef]
- Konstantinidis, I.T.; Warshaw, A.L.; Allen, J.N.; Blaszkowsky, L.S.; Castillo, C.F.; Deshpande, V.; Hong, T.S.; Kwak, E.L.; Lauwers, G.Y.; Ryan, D.P.; et al. Pancreatic ductal adenocarcinoma: Is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann. Surg. 2013, 257, 731–736. [Google Scholar] [CrossRef]
- Matters, G.L.; Cooper, T.K.; McGovern, C.O.; Gilius, E.L.; Liao, J.; Barth, B.M.; Kester, M.; Smith, J.P. Cholecystokinin mediates progression and metastasis of pancreatic cancer associated with dietary fat. Dig. Dis. Sci. 2014, 59, 1180–1191. [Google Scholar] [CrossRef]
- Osborne, N.; Sundseth, R.; Gay, M.D.; Cao, H.; Tucker, R.D.; Nadella, S.; Wang, S.; Liu, X.; Kroemer, A.; Sutton, L.; et al. Vaccine against gastrin, a polyclonal antibody stimulator, decreases pancreatic cancer metastases. Am. J. Physiol Gastrointest Liver Physiol. 2019, 317, G682–G693. [Google Scholar] [CrossRef]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Liposomal siRNA nanocarriers for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 110–116. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Soiza, R.L.; Scicluna, C.; Thomson, E.C. Efficacy and safety of COVID-19 vaccines in older people. Age Ageing. 2021, 50, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Dudley, M.Z.; Chen, X.; Bai, X.; Dong, K.; Zhuang, T.; Salmon, D.; Yu, H. Evaluation of the safety profile of COVID-19 vaccines: A rapid review. BMC Med. 2021, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Strain | C57BL/6 | C57BL/6 | KC | KC | KC |
|---|---|---|---|---|---|
| Treatment | Control | GAST siRNA | Control | Scrambled siRNA | MuKras siRNA |
| Blood Test/(Range) | |||||
| Alkaline Phosphatase | 56 ± 12 | 29 ± 5 | 76 ± 7 | 77 ± 8 | 74 ± 6 |
| (9.6–218.85 U/L) | |||||
| Alanine aminotransferase | 26 ± 6 | 13 ± 2 | 38 ± 9 | 60 ± 11 | 55 ±17 |
| (15.00–80.10 U/L) | |||||
| Aspartate aminotransferase | 159 ± 24 | 167 ± 52 | 156.75 | 122 ± 39 | 165 ± 22 |
| (33.95–268.47 U/L) | |||||
| Albumin | 3.2 ± 0.1 | 2.5 ± 0.2 | 3.3 ± 0.1 | 3.3 ± 0.2 | 3.2 ± 0.2 |
| (1.92–4.11 g/dL) | |||||
| Bilirubin | 0.3 ± 0.04 | 0.3 ± 0.05 | 0.33 ± 0.08 | 0.35 ± 0.09 | 0.3 ± 0.04 |
| (0.17–0.53 mg/dL) | |||||
| Calcium | 10.9 ± 0.6 | 10.9 ± 0.3 | 11.2 ± 0.2 | 11.5 ± 0.2 | 11.4 ± 0.4 |
| (8.28–12.27 mg/dL) | |||||
| Creatinine | 0.4 ± 0.03 | 0.3 ± 0.07 | 0.4 ± 0.04 | 0.4 ± 0.01 | 0.4 ± 0.04 |
| (0.12–0.43 mg/dL) | |||||
| BUN | 24 ± 2.7 | 30 ± 2.6 | 25 ± 2.1 | 25 ± 4.1 | 25 ± 2.1 |
| (9.42–31.53 mg/dL) | |||||
| Amylase | 529 ± 34 | 533 ± 199 | 853 ± 126 | 1274 ± 503 | 1254 ± 323 |
| (351-1563 U/L) | |||||
| Sodium | 141 ± 0.6 | 141 ± 0.4 | 141 ± 0.6 | 142 ± 0.8 | 143 ± 0.6 |
| (124.8–160.3 mmol/L) | |||||
| Potassium | 4 ± 0.06 | 4 ± 0.06 | 4.1 ± 0.04 | 4.2 ± 0.09 | 4.2 ± 0.04 |
| (2.17–8.18 mmol/L) | |||||
| Chloride | 106 ± 1.6 | 105 ± 2.3 | 109 ± 1.0 | 106 ± 2.6 | 108 ± 0.8 |
| (99.96–121.75 mmol/L) |
| Mouse Strain | Experiment | Treatments (N) | Objective | Sex |
|---|---|---|---|---|
| Athymic nude | Orthotopic Human AsPC-1 | PBS control (7), | Delivering 2 payloads. Measuring metastases | Female |
| 106 cells/mouse | NPs: Scrambled (7), GAST, (8) | Dose 240 nM TIW | ||
| muKRAS (8), Combination siRNA (8) | ||||
| Athymic nude | Orthotopic Human PANC-1 × 106 cells/mouse | PBS control (8) | Survival study, large tumor burden & | Female |
| NP: GAST siRNA (10) | Metastases; Dose 480 nM TIW | |||
| C57BL/6 | Orthotopic mouse mT3 | PBS Control (10), NPs: Scrambled (10), Combination muKras and WT- Kras siRNA (10) | Ki67 proliferation | Female |
| 100,000 cells/mouse | Survival | |||
| Deliver 2 payloads | ||||
| Dose 240 nM TIW | ||||
| Transgenic | P48-Cre/LSL-KrasG12D/+ | Control (no RX, 8), NPs: Scrambled siRNA (8), muKras (8) | Prevent PanINs | Males and females |
| Dose 480 nM BIW, long-term safety | ||||
| C57BL/6 | Subcutaneous mT3 | PBS Control (4) | Safety short term and off target toxicity | Males and females |
| Scrambled siRNA NP (4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.P.; Chen, W.; Shivapurkar, N.; Gerber, M.; Tucker, R.D.; Kallakury, B.; Dasa, S.S.K.; Kularatne, R.N.; Stern, S.T. Target-Specific Nanoparticle Polyplex Down-Regulates Mutant Kras to Prevent Pancreatic Carcinogenesis and Halt Tumor Progression. Int. J. Mol. Sci. 2023, 24, 752. https://doi.org/10.3390/ijms24010752
Smith JP, Chen W, Shivapurkar N, Gerber M, Tucker RD, Kallakury B, Dasa SSK, Kularatne RN, Stern ST. Target-Specific Nanoparticle Polyplex Down-Regulates Mutant Kras to Prevent Pancreatic Carcinogenesis and Halt Tumor Progression. International Journal of Molecular Sciences. 2023; 24(1):752. https://doi.org/10.3390/ijms24010752
Chicago/Turabian StyleSmith, Jill P., Wenqiang Chen, Narayan Shivapurkar, Monica Gerber, Robin D. Tucker, Bhaskar Kallakury, Siva Sai Krishna Dasa, Ruvanthi N. Kularatne, and Stephan T. Stern. 2023. "Target-Specific Nanoparticle Polyplex Down-Regulates Mutant Kras to Prevent Pancreatic Carcinogenesis and Halt Tumor Progression" International Journal of Molecular Sciences 24, no. 1: 752. https://doi.org/10.3390/ijms24010752
APA StyleSmith, J. P., Chen, W., Shivapurkar, N., Gerber, M., Tucker, R. D., Kallakury, B., Dasa, S. S. K., Kularatne, R. N., & Stern, S. T. (2023). Target-Specific Nanoparticle Polyplex Down-Regulates Mutant Kras to Prevent Pancreatic Carcinogenesis and Halt Tumor Progression. International Journal of Molecular Sciences, 24(1), 752. https://doi.org/10.3390/ijms24010752
{kind=link}