Anammox Bacterial S-Adenosyl-l-Methionine Dependent Methyltransferase Crystal Structure and Its Interaction with Acyl Carrier Proteins

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
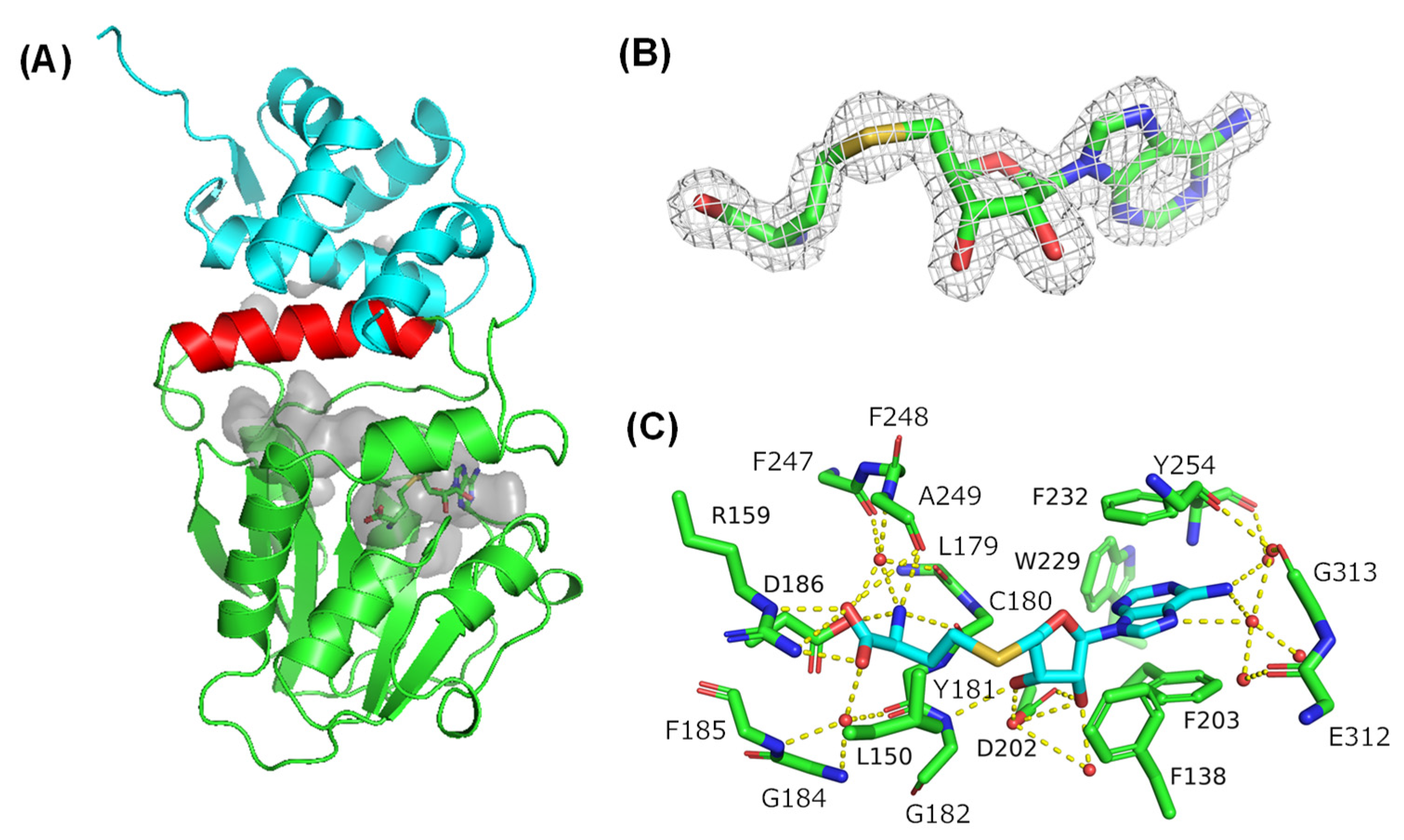
2.1. Overall Structure of Amx MT1 and Cosubstrate Binding Mode
2.2. Comparison with Other SAM-MTs
2.3. ACP/C-MT and AmxACP/AmxMT1 Complex Models
2.4. AmxMT1 Pull-Down Assay
2.5. Potential Substrate Binding Mode and Reaction Mechanisms of AmxMT1
2.6. Conclusions
3. Materials and Methods
3.1. Protein Production and Purification
3.2. Crystallization, Data Collection, and Structure Determination of AmxMT1
3.3. Preparation of ACP-Immobilized Sepharose and Pull-Down Assay
3.4. Potential Substrate Docking Simulation of AmxMT1
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demendoza, D.; Cronan, J.E. Thermal regulation of membrane lipid fluidity in bacteria. Trends Biochem. Sci. 1983, 8, 49–52. [Google Scholar] [CrossRef]
- van de Vossenberg, J.; Driessen, A.J.M.; da Costa, M.S.; Konings, W.N. Homeostasis of the membrane proton permeability in Bacillus subtilis grown at different temperatures. Biochim. Biophys. Acta-Biomembr. 1999, 1419, 97–104. [Google Scholar] [CrossRef]
- Van de Graaf, A.A.; Mulder, A.; de Bruijn, P.E.T.E.R.; Jetten, M.S.; Robertson, L.A.; Kuenen, J.G. Anaerobic oxidation of ammonium is a biologically mediated process. Appl. Environ. Microbiol. 1995, 61, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Kartal, B.; de Almeida, N.M.; Maalcke, W.J.; Op den Camp, H.J.; Jetten, M.S.; Keltjens, J.T. How to make a living from anaerobic ammonium oxidation. FEMS Microbiol. Rev. 2013, 37, 428–461. [Google Scholar] [CrossRef]
- Kartal, B.; Keltjens, J.T. Anammox Biochemistry: A Tale of Heme c Proteins. Trends Biochem. Sci. 2016, 41, 998–1011. [Google Scholar] [CrossRef]
- Peeters, S.H.; van Niftrik, L. Trending topics and open questions in anaerobic ammonium oxidation. Curr. Opin. Chem. Biol. 2019, 49, 45–52. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, N.M.; Neumann, S.; Mesman, R.J.; Ferousi, C.; Keltjens, J.T.; Jetten, M.S.; Kartal, B.; van Niftrik, L. Immunogold localization of key metabolic enzymes in the anammoxosome and on the tubule-like structures of Kuenenia stuttgartiensis. J. Bacteriol. 2015, 197, 2432–2441. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Wessels, H.J.; Rijpstra, W.I.C.; Sinninghe Damsté, J.S.; Kartal, B.; Jetten, M.S.; van Niftrik, L. Isolation and characterization of a prokaryotic cell organelle from the anammox bacterium Kuenenia stuttgartiensis. Mol. Microbiol. 2014, 94, 794–802. [Google Scholar] [CrossRef]
- Sinninghe Damsté, J.S.; Strous, M.; Rijpstra, W.I.C.; Hopmans, E.C.; Geenevasen, J.A.; Van Duin, A.C.; Van Niftrik, L.A.; Jetten, M.S. Linearly concatenated cyclobutane lipids form a dense bacterial membrane. Nature 2002, 419, 708–712. [Google Scholar] [CrossRef]
- Moss, F.R., 3rd; Shuken, S.R.; Mercer, J.A.; Cohen, C.M.; Weiss, T.M.; Boxer, S.G.; Burns, N.Z. Ladderane phospholipids form a densely packed membrane with normal hydrazine and anomalously low pro-ton/hydroxide permeability. Proc. Natl. Acad. Sci. USA 2018, 115, 9098–9103. [Google Scholar] [CrossRef]
- Zhang, L.; Narita, Y.; Gao, L.; Ali, M.; Oshiki, M.; Okabe, S. Maximum specific growth rate of anammox bacteria revisited. Water Res. 2017, 116, 296–303. [Google Scholar] [CrossRef]
- Mascitti, V.; Corey, E.J. Total synthesis of (+/−)-pentacycloanammoxic acid. J. Am. Chem. Soc. 2004, 126, 15664–15665. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.A.; Cohen, C.M.; Shuken, S.R.; Wagner, A.M.; Smith, M.W.; Moss, F.R., III; Smith, M.D.; Vahala, R.; Gonzalez-Martinez, A.; Boxer, S.G.; et al. Chemical Synthesis and Self-Assembly of a Ladderane Phospholipid. J. Am. Chem. Soc. 2016, 138, 15845–15848. [Google Scholar] [CrossRef] [PubMed]
- Rattray, J.E.; Strous, M.; Op den Camp, H.J.; Schouten, S.; Jetten, M.S.; Damsté, J.S.S. A comparative genomics study of genetic products potentially encoding ladderane lipid biosynthesis. Biol. Direct 2009, 4, 16. [Google Scholar] [CrossRef]
- Javidpour, P.; Deutsch, S.; Mutalik, V.K.; Hillson, N.J.; Petzold, C.J.; Keasling, J.D.; Beller, H.R. Investigation of proposed ladderane biosynthetic genes from anammox bacteria by heterologous expression in E-coli. PLoS ONE 2016, 11, e0151087. [Google Scholar] [CrossRef] [PubMed]
- Rattray, J.E.; van de Vossenberg, J.; Hopmans, E.C.; Kartal, B.; van Niftrik, L.; Rijpstra, W.I.C.; Strous, M.; Jetten, M.S.; Schouten, S.; Damsté, J.S.S. Ladderane lipid distribution in four genera of anammox bacteria. Arch. Microbiol. 2008, 190, 51–66. [Google Scholar] [CrossRef]
- Martin, J.L.; McMillan, F.M. SAM (dependent) I am: The S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 2002, 12, 783–793. [Google Scholar] [CrossRef]
- Laurino, P.; Tóth-Petróczy, Á.; Meana-Pañeda, R.; Lin, W.; Truhlar, D.G.; Tawfik, D.S. An Ancient Fingerprint Indicates the Common Ancestry of Rossmann-Fold Enzymes Utilizing Different Ribose-Based Cofactors. PLoS Biol. 2016, 14, e1002396. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Tsunematsu, Y.; Matsushita, T.; Hara, K.; Hashimoto, H.; Tang, Y.; Watanabe, K. Functional and Structural Analyses of trans C-Methyltransferase in Fungal Polyketide Biosynthesis. Biochemistry 2019, 58, 3933–3937. [Google Scholar] [CrossRef] [PubMed]
- Skiba, M.A.; Sikkema, A.P.; Fiers, W.D.; Gerwick, W.H.; Sherman, D.H.; Aldrich, C.C.; Smith, J.L. Domain Organization and Active Site Architecture of a Polyketide Synthase C-methyltransferase. ACS Chem. Biol. 2016, 11, 3319–3327. [Google Scholar] [CrossRef]
- Storm, P.A.; Herbst, D.A.; Maier, T.; Townsend, C.A. Functional and structural analysis of programmed C-methylation in the biosynthesis of the fungal polyketide citrinin. Cell Chem. Biol. 2017, 24, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, K.; Jackowski, S.; Rock, C.O.; Cronan, J.E. Regulation of fatty-acid biosynthesis in escherichia-coli. Microbiol. Rev. 1993, 57, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.C.; Lee, D.J.; Jackson, D.R.; Schaub, A.J.; Beld, J.; Barajas, J.F.; Hale, J.J.; Luo, R.; Burkart, M.D.; Tsai, S.C. Molecular basis for interactions between an acyl carrier protein and a ketosynthase. Nat. Chem. Biol. 2019, 15, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 2022, 31, 19. [Google Scholar] [CrossRef]
- Kim, Y.M.; Prestegard, J.H. Refinement of the NMR structures for acyl carrier protein with scalar coupling data. Proteins 1990, 8, 377–385. [Google Scholar] [CrossRef]
- Dietl, A.; Barends, T.R.M. Dynamics in an unusual acyl carrier protein from a ladderane lipid-synthesizing organism. Proteins 2022, 90, 73–82. [Google Scholar] [CrossRef]
- Parris, K.D.; Lin, L.; Tam, A.; Mathew, R.; Hixon, J.; Stahl, M.; Fritz, C.C.; Seehra, J.; Somers, W.S. Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Struct. Fold. Des. 2000, 8, 883–895. [Google Scholar] [CrossRef]
- Chen, S.S.; Tantillo, D.J. Potential for Ladderane (Bio)synthesis from Oligo-Cyclopropane Precursors. ACS Omega 2020, 5, 26134–26140. [Google Scholar] [CrossRef]
- Grogan, D.W.; Cronan, J.E. Cyclopropane ring formation in membrane lipids of bacteria. Microbiol. Mol. Biol. Rev. 1997, 61, 429–441. [Google Scholar]
- Glickman, M.S.; Cox, J.S.; Jacobs, W.R. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell 2000, 5, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.B.; Grant, R.A.; Sauer, R.T. Structural and Functional Analysis of E-coli Cyclopropane Fatty Acid Synthase. Structure 2018, 26, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.L.; Pan, C.L.; Wang, Q.H. Crystal structure of bacterial cyclopropane-fatty-acylphospholipid synthase with phospholipid. J. Biochem. 2019, 166, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Smith, C.V.; Glickman, M.S.; Jacobs, W.R.; Sacchettini, J.C. Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis. J. Biol. Chem. 2002, 277, 11559–11569. [Google Scholar] [CrossRef]
- Iwig, D.F.; Uchida, A.; Stromberg, J.A.; Booker, S.J. The activity of Escherichia coli cyclopropane fatty acid synthase depends on the presence of bicarbonate. J. Am. Chem. Soc. 2005, 127, 11612–11613. [Google Scholar] [CrossRef]
- Luo, Z.P. Selenourea: A convenient phasing vehicle for macromolecular X-ray crystal structures. Sci. Rep. 2016, 6, 6. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D 2019, 75, 861–877. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uegaki, T.; Takei, T.; Yamaguchi, S.; Fujiyama, K.; Sato, Y.; Hino, T.; Nagano, S. Anammox Bacterial S-Adenosyl-l-Methionine Dependent Methyltransferase Crystal Structure and Its Interaction with Acyl Carrier Proteins. Int. J. Mol. Sci. 2023, 24, 744. https://doi.org/10.3390/ijms24010744
Uegaki T, Takei T, Yamaguchi S, Fujiyama K, Sato Y, Hino T, Nagano S. Anammox Bacterial S-Adenosyl-l-Methionine Dependent Methyltransferase Crystal Structure and Its Interaction with Acyl Carrier Proteins. International Journal of Molecular Sciences. 2023; 24(1):744. https://doi.org/10.3390/ijms24010744
Chicago/Turabian StyleUegaki, Tesshin, Taisei Takei, Shuhei Yamaguchi, Keisuke Fujiyama, Yusuke Sato, Tomoya Hino, and Shingo Nagano. 2023. "Anammox Bacterial S-Adenosyl-l-Methionine Dependent Methyltransferase Crystal Structure and Its Interaction with Acyl Carrier Proteins" International Journal of Molecular Sciences 24, no. 1: 744. https://doi.org/10.3390/ijms24010744
APA StyleUegaki, T., Takei, T., Yamaguchi, S., Fujiyama, K., Sato, Y., Hino, T., & Nagano, S. (2023). Anammox Bacterial S-Adenosyl-l-Methionine Dependent Methyltransferase Crystal Structure and Its Interaction with Acyl Carrier Proteins. International Journal of Molecular Sciences, 24(1), 744. https://doi.org/10.3390/ijms24010744