
LUHMES Cells: Phenotype Refinement and Development of an MPP+-Based Test System for Screening Antiparkinsonian Drugs


Abstract
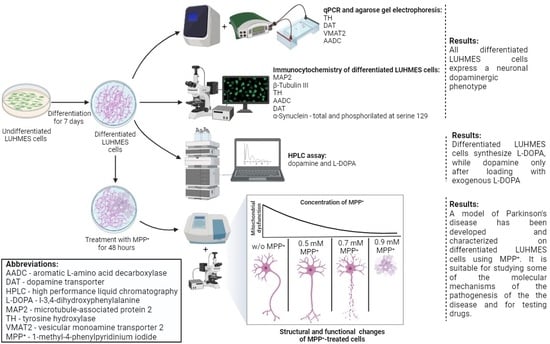
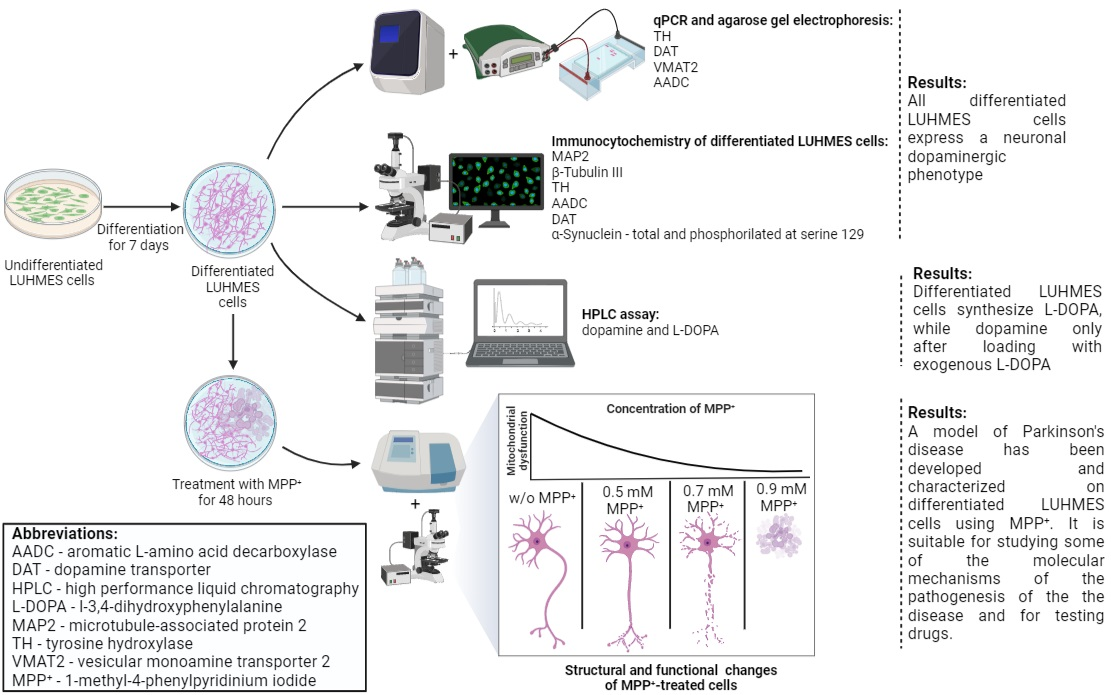
1. Introduction
2. Results
2.1. Specification of Differentiated LUHMES Cells
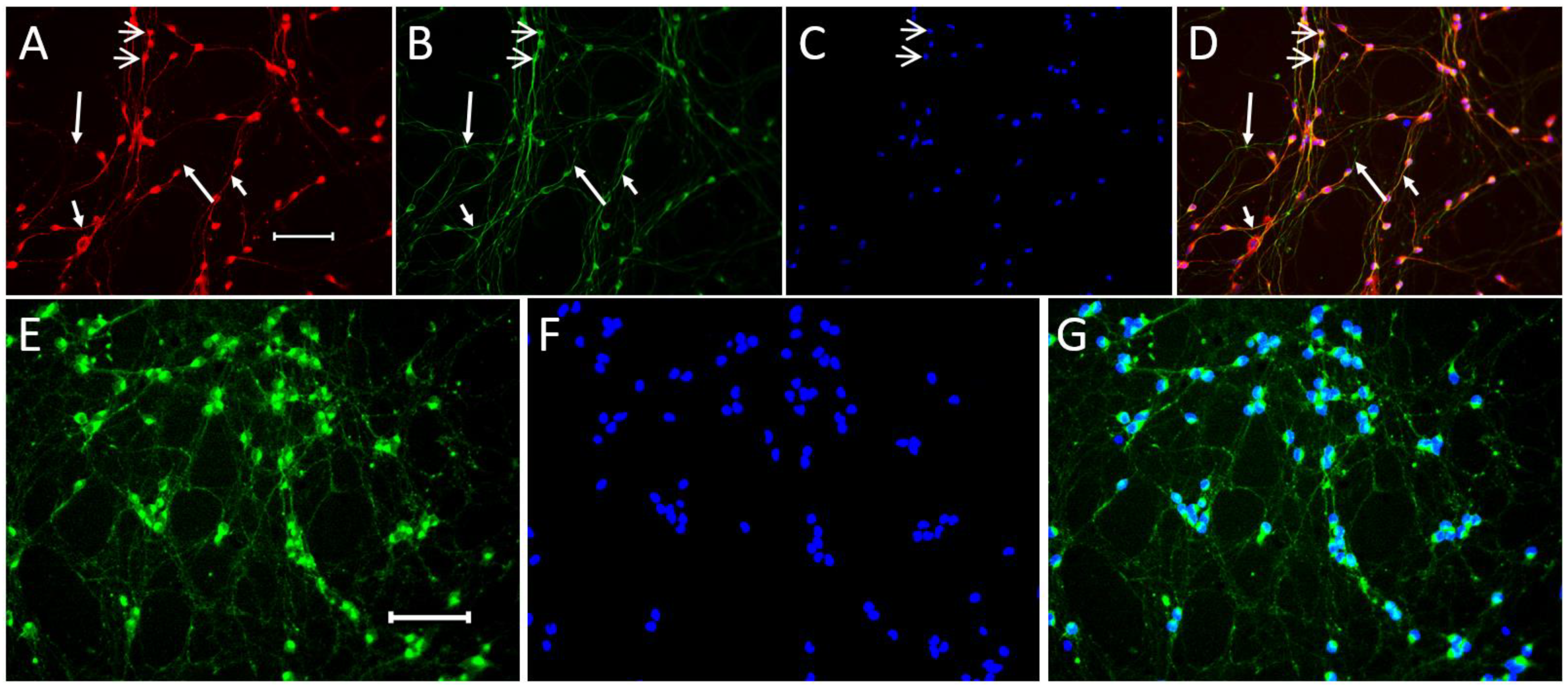
2.1.1. Expression of Neuronal Phenotype Proteins
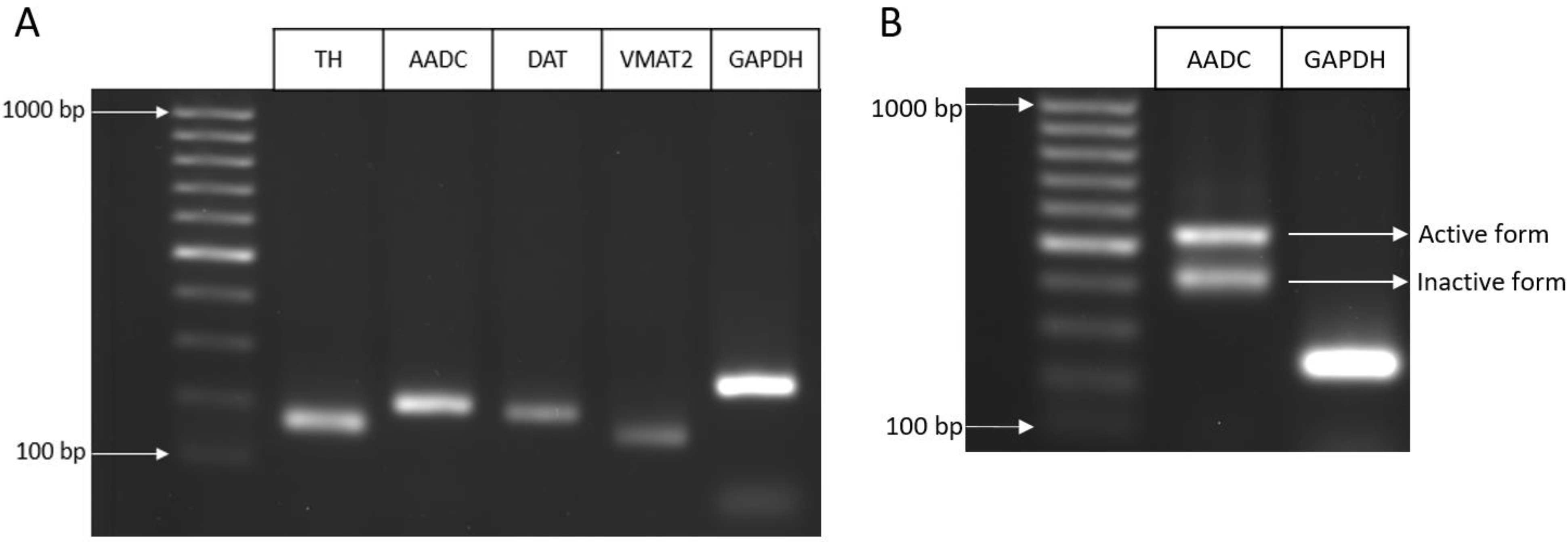
2.1.2. Expression of Genes for Proteins of the Dopaminergic Phenotype
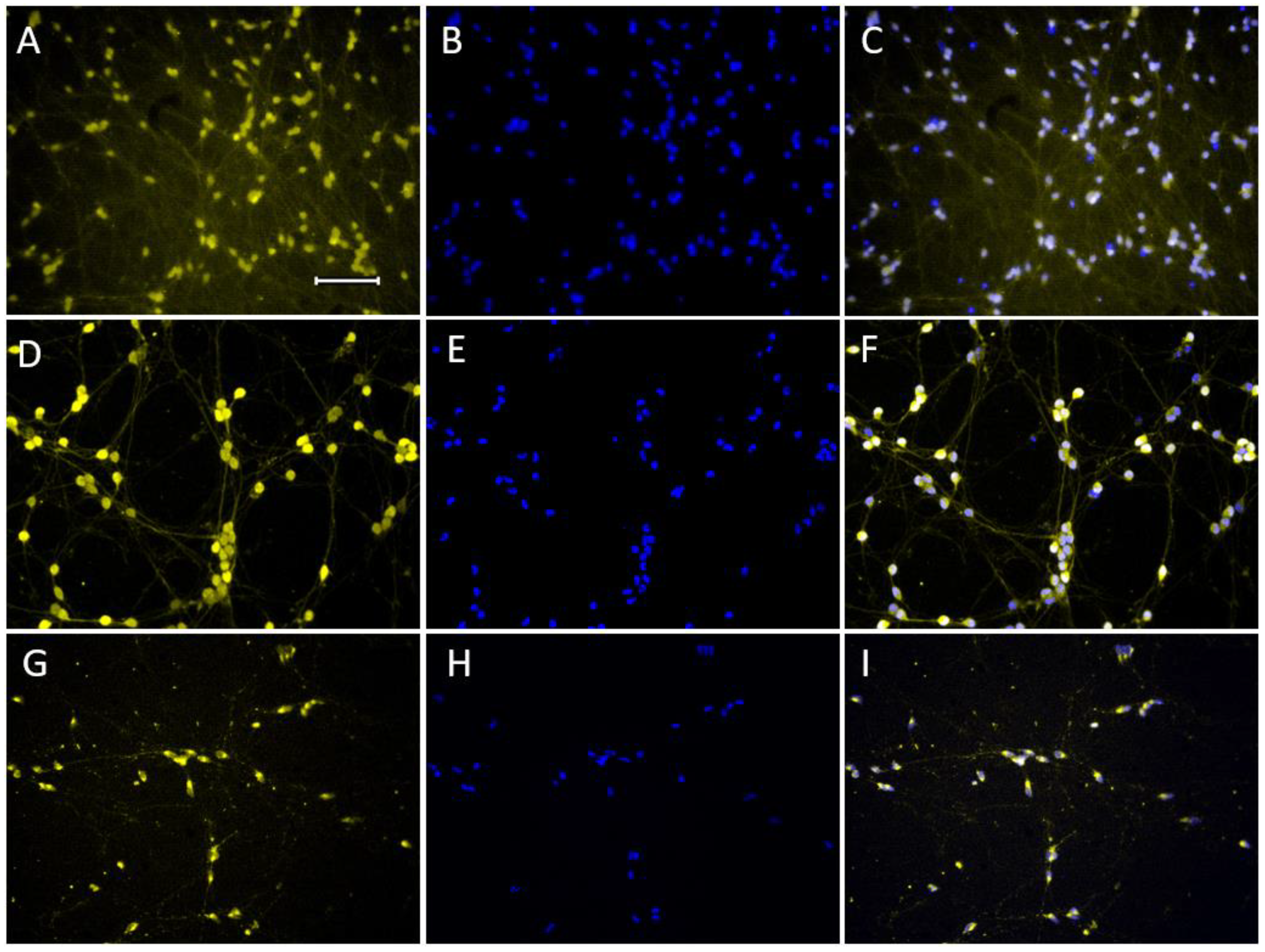
2.1.3. Expression of Dopaminergic Phenotype Proteins
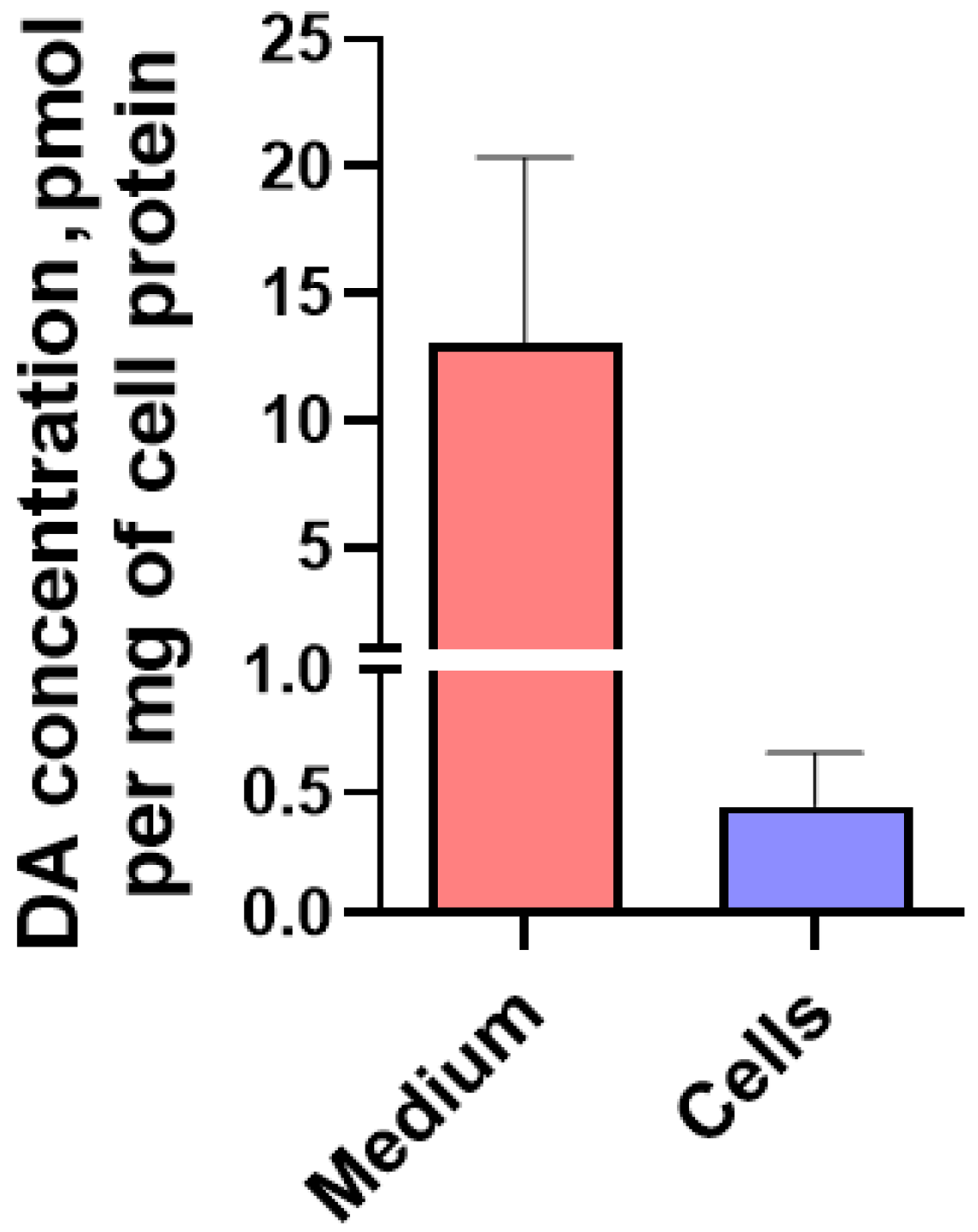
2.1.4. Content of L-3,4-dihydroxyphenylalanine, pyridoxal-5-phosphate and Dopamine in LUHMES Cells and in the Incubation Medium under Various Conditions
2.2. Modeling Parkinson’s Disease on LUHMES Cells Using 1-methyl-4-phenylpyridinium Iodide (MPP+)
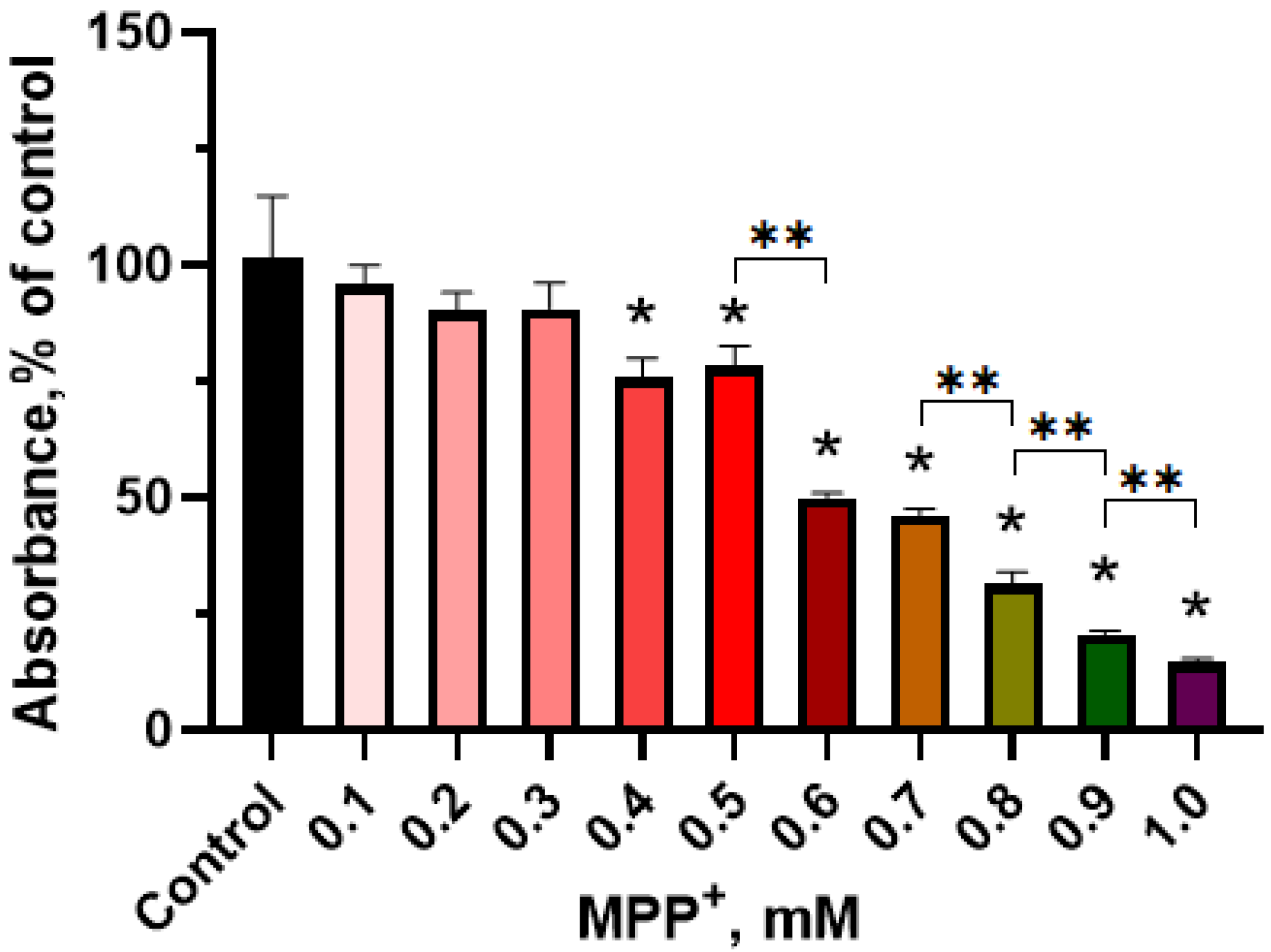
2.2.1. Evaluation of the Functional Activity of Mitochondria in LUHMES Cells Using the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Test when Exposed to 1-methyl-4-phenylpyridinium Iodide
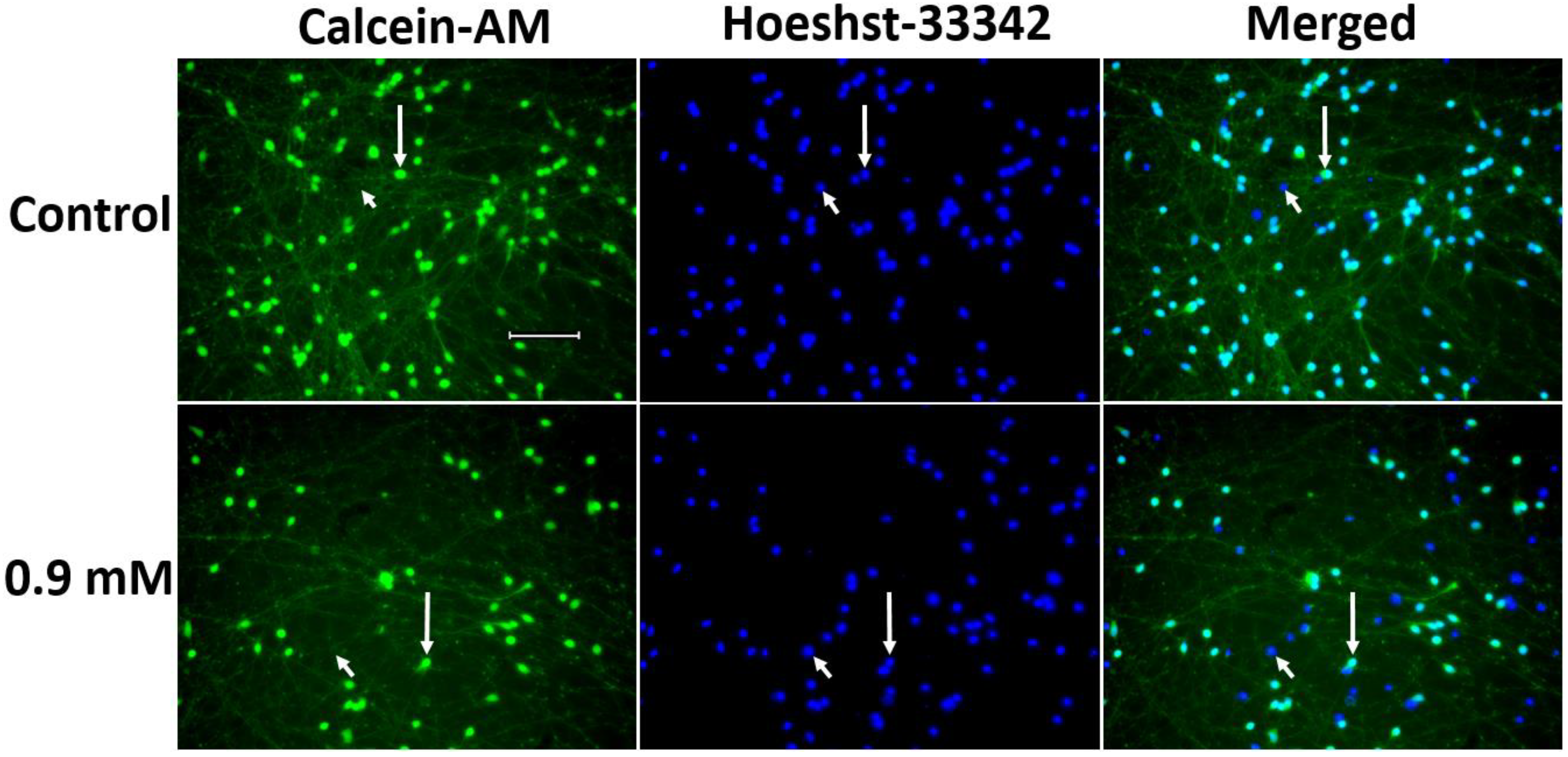
2.2.2. Evaluation of 1-methyl-4-phenylpyridinium Iodide-Induced Degeneration of LUHMES Cells
3. Discussion
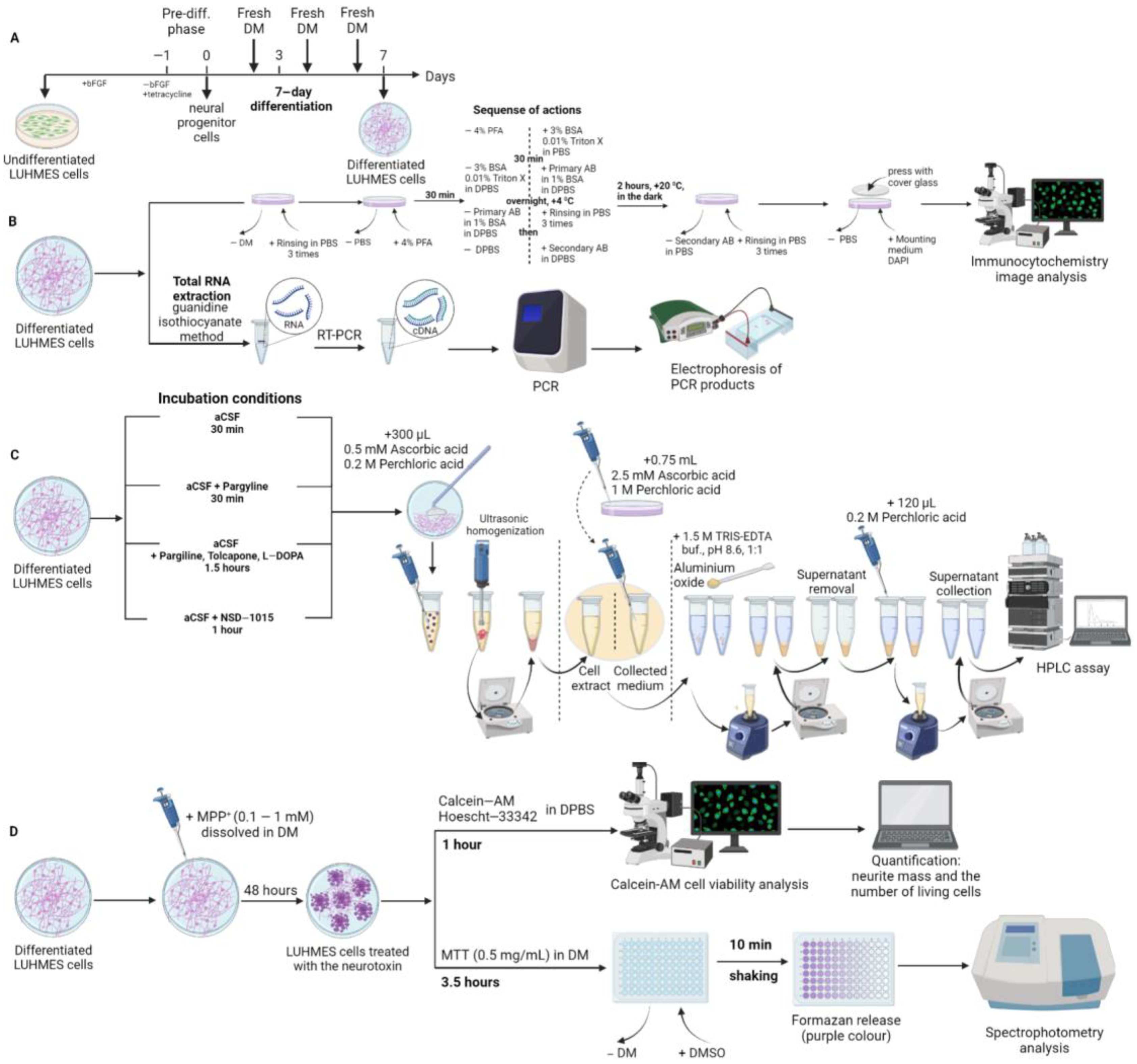
4. Materials and Methods
4.1. LUHMES Cell Cultivation
4.1.1. Preparing Plastic Dishes
4.1.2. Subculturing Undifferentiated Cells
4.1.3. Cell Differentiation
4.1.4. Experiments with LUHMES Cells
4.2. Cell Fixation and Immunostaining
4.3. High Performance Liquid Chromatography
4.4. Quantitative Real-Time Polymerase Chain Reaction and Agarose Gel Electrophoresis
4.5. Modeling of Parkinson’s Disease on LUHMES Cells Using 1-methyl-4-phenylpyridinium Iodide
4.5.1. Cell Survival Assessment
4.5.2. Evaluation of the Functional Activity of Mitochondria Using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide Test
4.6. Microscopy, Image Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AADC | aromatic L-amino acid decarboxylase |
| ATCC | American Type Culture Collection |
| DA | dopamine |
| DAergic | dopaminergic |
| DAPI | 4′,6-diamidino-2-phenylindole dihydrochloride |
| DAT | dopamine transporter |
| DPBS | Dulbecco’s phosphate-buffered saline without Ca2+ and Mg2+ |
| LCs | LUHMES cells |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| MAP2 | microtubule-associated protein 2 |
| MPP+ | 1-methyl-4-phenylpyridinium iodide |
| PCR | polymerase chain reaction |
| PD | Parkinson’s disease |
| TH | tyrosine hydroxylase |
| UKN | University of Konstanz |
| VMAT2 | vesicular monoamine transporter 2 |
References
- Agid, Y. Parkinson’s Disease: Pathophysiology. Lancet 1991, 337, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Picelli, A.; Tocco, P.; Federico, A.; Roncari, L.; Smania, N.; Zanette, G.; Tamburin, S. Pathophysiology of Motor Dysfunction in Parkinson’s Disease as the Rationale for Drug Treatment and Rehabilitation. Park. Dis. 2016, 2016, 9832839. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Rodriguez, E.; Vegas-Suarez, S.; Morera-Herreras, T.; De Deurwaerdere, P.; Miguelez, C. The Noradrenergic System in Parkinson’s Disease. Front. Pharmacol. 2020, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, L.Y.; Copersino, M.L.; Fang, Y.X.; Chen, Y.; Tian, J.; Deng, Y.; Shuai, Y.; Jin, J.; Lu, L. PET Imaging of Dopamine Transporter and Drug Craving during Methadone Maintenance Treatment and after Prolonged Abstinence in Heroin Users. Eur. J. Pharmacol. 2008, 579, 160–166. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.J.; Kawamata, H.; Ribich, S.; Hyman, B.T. Membrane Association and Protein Conformation of α-Synuclein in Intact Neurons. Effect of Parkinson’s Disease-Linked Mutations. J. Biol. Chem. 2000, 275, 8812–8816. [Google Scholar] [CrossRef] [PubMed]
- Hodaie, M.; Neimat, J.S.; Lozano, A.M. The Dopaminergic Nigrostriatal System and Parkinson’s Disease: Molecular Events in Development, Disease, and Cell Death, and New Therapeutic Strategies. Neurosurgery 2007, 60, 17–28. [Google Scholar] [CrossRef]
- Bezard, E.; Gross, C.E. Compensatory Mechanisms in Experimental and Human Parkinsonism: Towards a Dynamic Approach. Prog. Neurobiol. 1998, 55, 93–116. [Google Scholar] [CrossRef]
- Ugrumov, M. Development of Early Diagnosis of Parkinson’s Disease: Illusion or Reality? CNS Neurosci. Ther. 2020, 26, 997–1009. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, M. Cell-Based Assays for Parkinson’s Disease Using Differentiated Human LUHMES Cells. Acta Pharmacol. Sin. 2014, 35, 945–956. [Google Scholar] [CrossRef]
- Santos, N.A.G.; Martins, N.M.; Sisti, F.M.; Fernandes, L.S.; Ferreira, R.S.; Queiroz, R.H.C.; Santos, A.C. The Neuroprotection of Cannabidiol against MPP + -Induced Toxicity in PC12 Cells Involves TrkA Receptors, Upregulation of Axonal and Synaptic Proteins, Neuritogenesis, and Might Be Relevant to Parkinson’s Disease. Toxicol. Vitr. 2015, 30, 231–240. [Google Scholar] [CrossRef]
- Ramalingam, M.; Kim, S. The Neuroprotective Role of Insulin Against MPP + -Induced Parkinson’s Disease in Differentiated SH-SY5Y Cells. J. Cell. Biochem. 2016, 117, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH-SY5Y Cell Line in Parkinson’s Disease Research: A Systematic Review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Huh, Y.J.; Lee, Y.I. The Impairments of α-Synuclein and Mechanistic Target of Rapamycin in Rotenone-Induced SH-SY5Y Cells and Mice Model of Parkinson’s Disease. Front. Neurosci. 2019, 13, 1028. [Google Scholar] [CrossRef]
- Zhou, F.; Ju, J.; Fang, Y.; Fan, X.; Yan, S.; Wang, Q.; Wei, P.; Duan, F.; Miao, F.; Hu, Z.; et al. Salidroside Protected against MPP + -induced Parkinson’s Disease in PC12 Cells by Inhibiting Inflammation, Oxidative Stress and Cell Apoptosis. Biotechnol. Appl. Biochem. 2019, 66, 247–253. [Google Scholar] [CrossRef]
- Joshi, D.J.; Chitre, N.M.; Bansal, A.; Murnane, K.S.; D’Souza, M.J. Formulation and Characterization of Microcapsules Encapsulating PC12 Cells as a Prospective Treatment Approach for Parkinson’s Disease. AAPS PharmSciTech. 2021, 22, 149. [Google Scholar] [CrossRef]
- Lv, K.; Liu, Y.; Zheng, Y.; Dai, S.; Yin, P.; Miao, H. Long Non-coding RNA MALAT1 Regulates Cell Proliferation and Apoptosis via MiR-135b-5p/GPNMB Axis in Parkinson’s Disease Cell Model. Biol. Res. 2021, 54, 10. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.M.; Wrench, N.; Ryde, I.T.; Smith, A.M.; Seidler, F.J.; Slotkin, T.A. Silver Impairs Neurodevelopment: Studies in PC12 Cells. Environ. Health Perspect. 2010, 118, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Pouresmaeili-Babaki, E.; Esmaeili-Mahani, S.; Abbasnejad, M.; Ravan, H. Protective Effect of Neuropeptide Apelin-13 on 6-Hydroxydopamine-Induced Neurotoxicity in SH-SY5Y Dopaminergic Cells: Involvement of Its Antioxidant and Antiapoptotic Properties. Rejuvenation Res. 2018, 21, 162–167. [Google Scholar] [CrossRef]
- Martin, E.R.; Gandawijaya, J.; Oguro-Ando, A. A Novel Method for Generating Glutamatergic SH-SY5Y Neuron-like Cells Utilizing B-27 Supplement. Front. Pharmacol. 2022, 13, 943627. [Google Scholar] [CrossRef]
- Zhang, D.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Neuroprotective Effect of Protein Kinase Cδ Inhibitor Rottlerin in Cell Culture and Animal Models of Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2007, 322, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal Infection Triggers Parkinson’s Disease-like Symptoms in Pink1−/− Mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, R.; Kamrava, S.K.; Bagher, Z.; Farhadi, M.; Falah, M.; Moradi, F.; Boroujeni, M.E.; Soleimani, M.; Kamyab, A.; Komeili, A. Human Olfactory Stem Cells: As a Promising Source of Dopaminergic Neuron-like Cells for Treatment of Parkinson’s Disease. Neurosci. Lett. 2019, 696, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.V.; Larroquette, F.; Beitel, L.K.; Fon, E.A.; Durcan, T.M. One Step Into the Future: New IPSC Tools to Advance Research in Parkinson’s Disease and Neurological Disorders. J. Park. Dis. 2019, 9, 17. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized IPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Song, B.; Cha, Y.; Ko, S.; Jeon, J.; Lee, N.; Seo, H.; Park, K.J.; Lee, I.H.; Lopes, C.; Feitosa, M.; et al. Human Autologous IPSC–Derived Dopaminergic Progenitors Restore Motor Function in Parkinson’s Disease Models. J. Clin. Investig. 2020, 130, 904–920. [Google Scholar] [CrossRef]
- Lotharius, J. Progressive Degeneration of Human Mesencephalic Neuron-Derived Cells Triggered by Dopamine-Dependent Oxidative Stress Is Dependent on the Mixed-Lineage Kinase Pathway. J. Neurosci. 2005, 25, 6329–6342. [Google Scholar] [CrossRef]
- Schildknecht, S.; Pöltl, D.; Nagel, D.M.; Matt, F.; Scholz, D.; Lotharius, J.; Schmieg, N.; Salvo-Vargas, A.; Leist, M. Requirement of a Dopaminergic Neuronal Phenotype for Toxicity of Low Concentrations of 1-Methyl-4-Phenylpyridinium to Human Cells. Toxicol. Appl. Pharmacol. 2009, 241, 23–35. [Google Scholar] [CrossRef]
- Scholz, D.; Pöltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, Complete and Large-Scale Generation of Post-Mitotic Neurons from the Human LUHMES Cell Line. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef]
- Pöltl, D.; Schildknecht, S.; Karreman, C.; Leist, M. Uncoupling of ATP-Depletion and Cell Death in Human Dopaminergic Neurons. Neurotoxicology 2012, 33, 769–779. [Google Scholar] [CrossRef]
- Brüll, M.; Spreng, A.S.; Gutbier, S.; Loser, D.; Krebs, A.; Reich, M.; Kraushaar, U.; Britschgi, M.; Patsch, C.; Leist, M. Incorporation of stem cell-derived astrocytes into neuronal organoids to allow neuro-glial interactions in toxicological studies. ALTEX 2020, 37, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Gutbier, S.; May, P.; Berthelot, S.; Krishna, A.; Trefzer, T.; Behbehani, M.; Efremova, L.; Delp, J.; Gstraunthaler, G.; Waldmann, T.; et al. Major Changes of Cell Function and Toxicant Sensitivity in Cultured Cells Undergoing Mild, Quasi-Natural Genetic Drift. Arch. Toxicol. 2018, 92, 3487–3503. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The Costs of Using Unauthenticated, over-Passaged Cell Lines: How Much More Data Do We Need? Biotechniques 2007, 43, 575–586. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, K.L.; Harmon, S.; Moffat, M.; Uhland-Smith, A.; Wong, S. The Human Aromatic L-Amino Acid Decarboxylase Gene Can Be Alternatively Spliced To Generate Unique Protein Isoforms. J. Neurochem. 2002, 65, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-Inducing Neurotoxin, N-Methyl-4-Phenyl-1,2,3,6 -Tetrahydropyridine: Uptake of the Metabolite N-Methyl-4-Phenylpyridine by Dopamine Neurons Explains Selective Toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef]
- Bezard, E.; Gross, C.E.; Fournier, M.C.; Dovero, S.; Bloch, B.; Jaber, M. Absence of MPTP-Induced Neuronal Death in Mice Lacking the Dopamine Transporter. Exp. Neurol. 1999, 155, 268–273. [Google Scholar] [CrossRef]
- Choi, S.J.; Panhelainen, A.; Schmitz, Y.; Larsen, K.E.; Kanter, E.; Wu, M.; Sulzer, D.; Mosharov, E.V. Changes in Neuronal Dopamine Homeostasis Following 1-Methyl-4-Phenylpyridinium (MPP+) Exposure. J. Biol. Chem. 2015, 290, 6799–6809. [Google Scholar] [CrossRef]
- Akhtar, R.S.; Stern, M.B. New Concepts in the Early and Preclinical Detection of Parkinson’s Disease: Therapeutic Implications. Expert Rev. Neurother. 2012, 12, 1429–1438. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Cheong, S.L.; Federico, S.; Spalluto, G.; Klotz, K.N.; Pastorin, G. The Current Status of Pharmacotherapy for the Treatment of Parkinson’s Disease: Transition from Single-Target to Multitarget Therapy. Drug Discov. Today 2019, 24, 1769–1783. [Google Scholar] [CrossRef]
- Cooper, O.; Hargus, G.; Deleidi, M.; Blak, A.; Osborn, T.; Marlow, E.; Lee, K.; Levy, A.; Perez-Torres, E.; Yow, A.; et al. Differentiation of Human ES and Parkinson’s Disease IPS Cells into Ventral Midbrain Dopaminergic Neurons Requires a High Activity Form of SHH, FGF8a and Specific Regionalization by Retinoic Acid. Mol. Cell. Neurosci. 2010, 45, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Harischandra, D.S.; Rokad, D.; Ghaisas, S.; Verma, S.; Robertson, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Enhanced Differentiation of Human Dopaminergic Neuronal Cell Model for Preclinical Translational Research in Parkinson’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165533. [Google Scholar] [CrossRef]
- Lotharius, J.; Barg, S.; Wiekop, P.; Lundberg, C.; Raymon, H.K.; Brundin, P. Effect of Mutant α-Synuclein on Dopamine Homeostasis in a New Human Mesencephalic Cell Line. J. Biol. Chem. 2002, 277, 38884–38894. [Google Scholar] [CrossRef] [PubMed]
- Gaceb, A.; Barbariga, M.; Paul, G. An In Vitro Partial Lesion Model of Differentiated Human Mesencephalic Neurons: Effect of Pericyte Secretome on Phenotypic Markers. J. Mol. Neurosci. 2020, 70, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Binder, L.; Payne, M.; Bender, P.; Rebhun, L.; Steward, O. Differential Subcellular Localization of Tubulin and the Microtubule- Associated Protein MAP2 in Brain Tissue as Revealed by Immunocytochemistry with Monoclonal Hybridoma Antibodies. J. Neurosci. 1984, 4, 394–410. [Google Scholar] [CrossRef]
- Garner, C.C.; Tucker, R.P.; Matus, A. Selective Localization of Messenger RNA for Cytoskeletal Protein MAP2 in Dendrites. Nature 1988, 336, 674–677. [Google Scholar] [CrossRef]
- Südhof, T.C. The Molecular Machinery of Neurotransmitter Release (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 12696–12717. [Google Scholar] [CrossRef]
- Beyene, A.G.; McFarlane, I.R.; Pinals, R.L.; Landry, M.P. Stochastic Simulation of Dopamine Neuromodulation for Implementation of Fluorescent Neurochemical Probes in the Striatal Extracellular Space. ACS Chem. Neurosci. 2017, 8, 2275–2289. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Spillantini, M.G. Alpha-Synuclein Dysfunction in Lewy Body Diseases. Mov. Disord. 2005, 20, S37–S44. [Google Scholar] [CrossRef]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 12. [Google Scholar] [CrossRef]
- Yan, R.; Li, Y.; Müller, J.; Zhang, Y.; Singer, S.; Xia, L.; Zhong, X.; Gertsch, J.; Altmann, K.H.; Zhou, Q. Mechanism of Substrate Transport and Inhibition of the Human LAT1-4F2hc Amino Acid Transporter. Cell Discov. 2021, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Gjedde, A.; Reith, J.; Dyve, S.; Léger, G.; Guttman, M.; Diksic, M.; Evans, A.; Kuwabara, H. Dopa Decarboxylase Activity of the Living Human Brain. Proc. Natl. Acad. Sci. USA 1991, 88, 2721–2725. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Sumi-Ichinose, C.; Ohye, T.; Hagino, Y.; Fujita, K.; Nagatsu, T. Tissue-Specific Alternative Splicing of the First Exon Generates Two Types of MRNAs in Human Aromatic L-Amino Acid Decarboxylase. Biochemistry 1992, 31, 11546–11550. [Google Scholar] [CrossRef]
- Blechingberg, J.; Holm, I.E.; Johansen, M.G.; Børglum, A.D.; Nielsen, A.L. Aromatic L-Amino Acid Decarboxylase Expression Profiling and Isoform Detection in the Developing Porcine Brain. Brain Res. 2010, 1308, 1–13. [Google Scholar] [CrossRef]
- Lee, N.C.; Shieh, Y.D.; Chien, Y.H.; Tzen, K.Y.; Yu, I.S.; Chen, P.W.; Hu, M.H.; Hu, M.; Muramatsu, S.; Ichinose, H.; et al. Regulation of the Dopaminergic System in a Murine Model of Aromatic L-Amino Acid Decarboxylase Deficiency. Neurobiol. Dis. 2013, 52, 177–190. [Google Scholar] [CrossRef]
- Choi, H.K.; Won, L.; Roback, J.D.; Wainer, B.H.; Heller, A. Specific Modulation of Dopamine Expression in Neuronal Hybrid Cells by Primary Cells from Different Brain Regions. Proc. Natl. Acad. Sci. USA 1992, 89, 8943–8947. [Google Scholar] [CrossRef]
- Surkov, S.A.; Mingazov, E.R.; Blokhin, V.E.; Sturova, A.I.; Gretskaya, N.M.; Zinchenko, G.N.; Bezuglov, V.V.; Ugrumov, M.V. The Neuroprotective Effect of N-Docosahexaenoyldopamine on Degenerating Dopaminergic Neurons of the Mesencephalon. Biol. Bull. 2020, 47, 466–473. [Google Scholar] [CrossRef]
- Herkenham, M.; Little, M.D.; Bankiewicz, K.; Yang, S.C.; Markey, S.P.; Johannessen, J.N. Selective Retention of MPP+ within the Monoaminergic Systems of the Primate Brain Following MPTP Administration: An in Vivo Autoradiographic Study. Neuroscience 1991, 40, 133–158. [Google Scholar] [CrossRef]
- Fallon, J.; Matthews, R.T.; Hyman, B.T.; Beal, M.F. MPP+ Produces Progressive Neuronal Degeneration Which Is Mediated by Oxidative Stress. Exp. Neurol. 1997, 144, 193–198. [Google Scholar] [CrossRef]
- Nagatsu, T.; Nagatsu, I. Tyrosine Hydroxylase (TH), Its Cofactor Tetrahydrobiopterin (BH4), Other Catecholamine-Related Enzymes, and Their Human Genes in Relation to the Drug and Gene Therapies of Parkinson’s Disease (PD): Historical Overview and Future Prospects. J. Neural Transm. 2016, 123, 1255–1278. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Zuk, R.; Gazawi, H.; Ljubuncic, P. Dopamine Toxicity Involves Mitochondrial Complex I Inhibition: Implications to Dopamine-Related Neuropsychiatric Disorders. Biochem. Pharmacol. 2004, 67, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, Oxidative Stress and Protein–Quinone Modifications in Parkinson’s and Other Neurodegenerative Diseases. Angew. Chem. Int. Ed. 2019, 58, 6512–6527. [Google Scholar] [CrossRef]
- Smirnova, L.; Harris, G.; Delp, J.; Valadares, M.; Pamies, D.; Hogberg, H.T.; Waldmann, T.; Leist, M.; Hartung, T. A LUHMES 3D Dopaminergic Neuronal Model for Neurotoxicity Testing Allowing Long-Term Exposure and Cellular Resilience Analysis. Arch. Toxicol. 2016, 90, 2725–2743. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.R.; Tam, N.W.; Teixeira, A.G.; Frampton, J.P. SH-SY5Y and LUHMES Cells Display Differential Sensitivity to MPP+, Tunicamycin, and Epoxomicin in 2D and 3D Cell Culture. Biotechnol. Prog. 2020, 36, e2942. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; Cummins, C.L.; Mangelsdorf, D.J.; Pesola, J.M.; Kramer, M.F. High-Throughput Real-Time Quantitative Reverse Transcription PCR. Curr. Protoc. Mol. Biol. 2006, 73, 15.8.1–15.8.28. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Marfell, B.J.; Waterhouse, N.J. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harb. Protoc. 2016, 9, 778–781. [Google Scholar] [CrossRef]
- Blokhin, V.; Lavrova, A.V.; Surkov, S.A.; Mingazov, E.R.; Gretskaya, N.M.; Bezuglov, V.V.; Ugrumov, M.V. A New Method for the Visualization of Living Dopaminergic Neurons and Prospects for Using It to Develop Targeted Drug Delivery to These Cells. Int. J. Mol. Sci. 2022, 23, 3678. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic of DAergic Phenotype | UKN Line (Literature Data) | ATCC Line (Literature Data) | ATCC Line (Our Data) |
|---|---|---|---|
| TH | mRNA: + + [32] Protein: + + [28,29] | mRNA: + + [32] Protein: + + [10]; +/− [32,42] | mRNA: + + Protein: + + |
| AADC | mRNA: + + [29] Protein: − − | mRNA: + + [10] Protein: − − | mRNA: + + Protein: + |
| DAT | mRNA: + + [32] Protein: + [32]. | mRNA: + + [32] Protein: + [32] | mRNA: + + Protein: + + |
| VMAT2 | mRNA: + + [32] Protein: + + [43,44] | mRNA: + + [32] Protein: − − | mRNA: + + Protein: + + |
| DA | + + [28,29] | + − [32] | + − |
| Type of Antibodies | Manufacturer/Cat. No. | Specification | Dilution |
|---|---|---|---|
| Primary | Abcam (Cambridge, UK), ab111468 | Rabbit anti-DAT, polyclonal | 1:100 |
| Primary | Abcam (Cambridge, UK), ab32454 | Rabbit anti-MAP2, polyclonal | 1:250 |
| Primary | SySy (Goettingen, Germany), 105011 | Mouse anti-Synaptotagmin 1, monoclonal | 1:200 |
| Primary | Sigma (St. Louis, MO, USA), AB1598P | Rabbit anti-VMAT2, polyclonal | 1:75 |
| Primary | R&D systems (Minneapolis, MN, USA), AF3564 | Goat anti-AADC, polyclonal | 1:80 |
| Primary | Santa Cruz (Dallas, TX, USA), sc-7011-R | Rabbit anti-α-Synuclein, polyclonal | 1:250 |
| Primary | Abcam (Cambridge, UK), ab7751 | Mouse anti-β-Tubulin III, monoclonal | 1:1500 |
| Primary | Invitrogen (Waltham, MA, USA), PA5-37740 | Rabbit anti-Phospho-α-Synuclein (Ser-129), polyclonal | 1:750 |
| Primary | Thermo Fisher (Waltham, MA, USA), 701949 | Rabbit anti-TH, polyclonal | 1:250 |
| Primary | Sigma (St. Louis, MO, USA), T1299 | Mouse anti-TH, monoclonal | 1:500 |
| Secondary | Invitrogen (Waltham, MA, USA), A10040 | Donkey anti-Rabbit, Alexa Fluor 546 | 1:700 |
| Secondary | Invitrogen (Waltham, MA, USA), A32766 | Donkey anti-Mouse, Alexa Fluor 488 | 1:1000 |
| Secondary | Invitrogen (Waltham, MA, USA), A21469 | Chicken anti-Goat, Alexa Fluor 647 | 1:1000 |
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| TH | CGGGCTGTCCTCCTA | AGCTTGTCCTTGGCGTCACT |
| DAT | CAGCCTGCAGACCACACTC | AGGCTCGCGGATACTGC |
| VMAT2 | CCGGGAATGCTACCAGAGAC | CGAGGCAAACAACAGACCAA |
| AADC | GGCTTCCCATGTCTTTCTCC | GGCTTTCCTACCACTTCTGATG |
| AADC to test alternative splicing | GCCACCAGCTTCTCCATGA (516) | AGAGGGAAGGAGATGGTGGATTA (719) |
| GAPDH | GGGGGAGCCAAAAGGGTCATCATCT | GAGGGGCCATCCACAGTCTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beliakov, S.V.; Blokhin, V.; Surkov, S.A.; Ugrumov, M.V. LUHMES Cells: Phenotype Refinement and Development of an MPP+-Based Test System for Screening Antiparkinsonian Drugs. Int. J. Mol. Sci. 2023, 24, 733. https://doi.org/10.3390/ijms24010733
Beliakov SV, Blokhin V, Surkov SA, Ugrumov MV. LUHMES Cells: Phenotype Refinement and Development of an MPP+-Based Test System for Screening Antiparkinsonian Drugs. International Journal of Molecular Sciences. 2023; 24(1):733. https://doi.org/10.3390/ijms24010733
Chicago/Turabian StyleBeliakov, Sergei V., Victor Blokhin, Sergey A. Surkov, and Michael V. Ugrumov. 2023. "LUHMES Cells: Phenotype Refinement and Development of an MPP+-Based Test System for Screening Antiparkinsonian Drugs" International Journal of Molecular Sciences 24, no. 1: 733. https://doi.org/10.3390/ijms24010733
APA StyleBeliakov, S. V., Blokhin, V., Surkov, S. A., & Ugrumov, M. V. (2023). LUHMES Cells: Phenotype Refinement and Development of an MPP+-Based Test System for Screening Antiparkinsonian Drugs. International Journal of Molecular Sciences, 24(1), 733. https://doi.org/10.3390/ijms24010733