
Polycystin-1 Is a Crucial Regulator of BIN1 Expression and T-Tubule Remodeling Associated with the Development of Dilated Cardiomyopathy

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
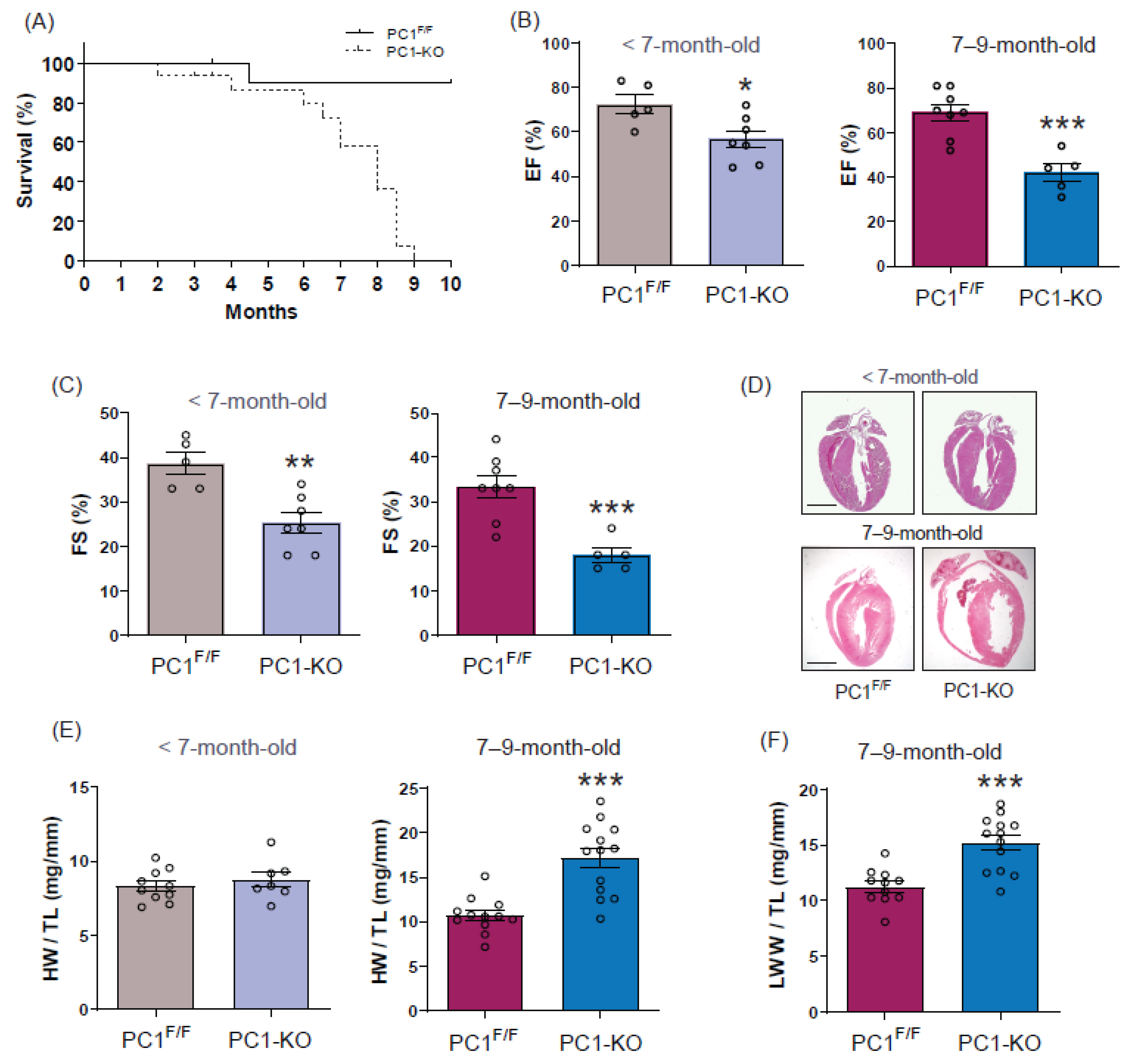
2.1. Sudden Death in Dilated Cardiomyopathy Is Associated with Loss of Polycystin-1 Expression
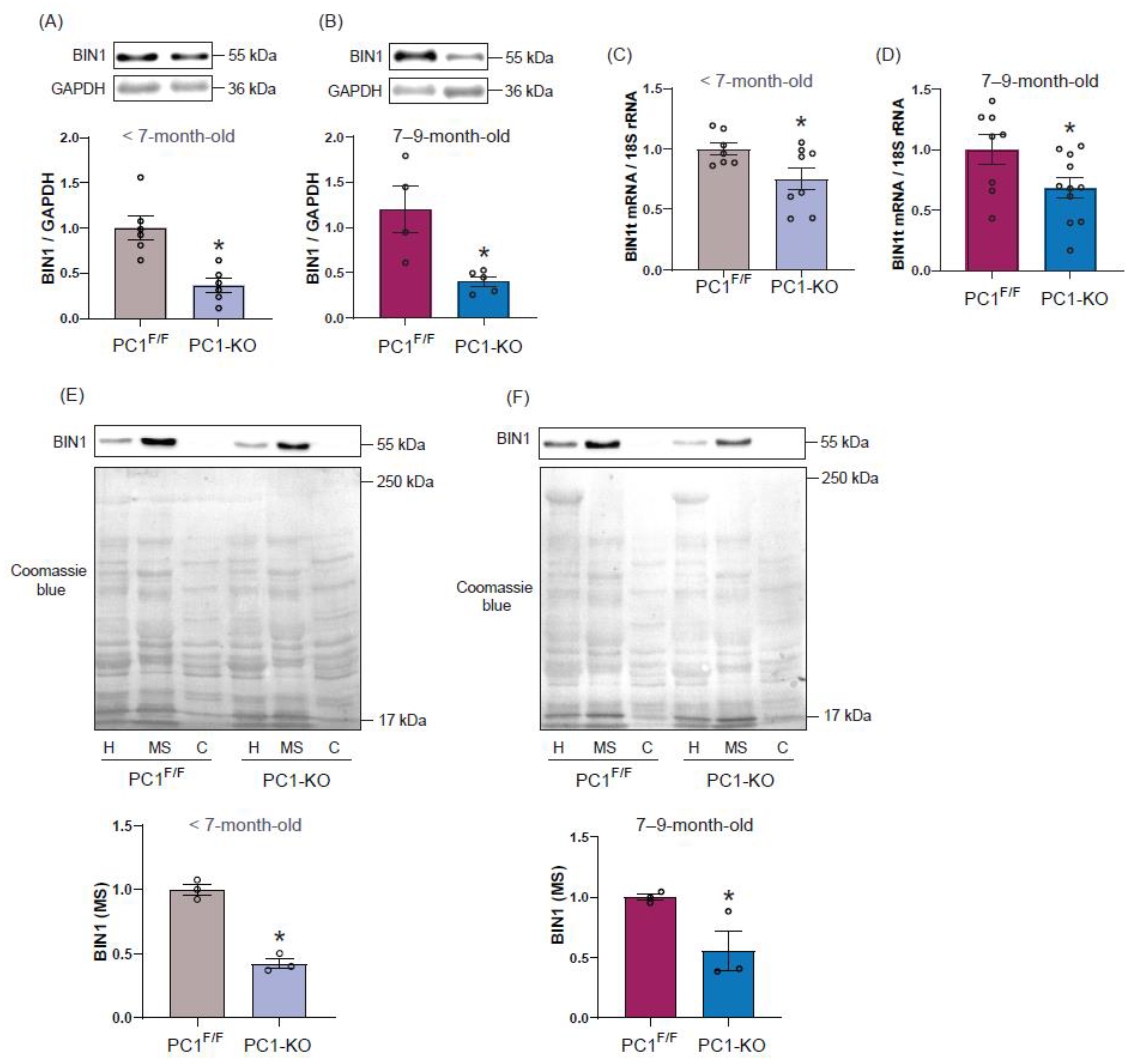
2.2. Polycystin-1 Regulates Cardiac BIN1 Expression
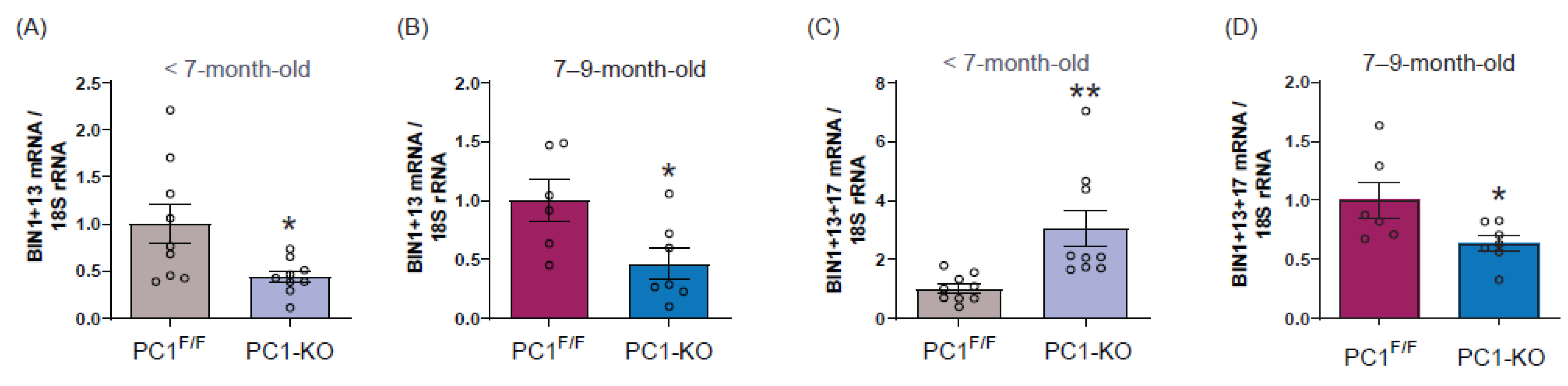
2.3. Cardiac BIN1 Isoform Expression Is Regulated by Polycystin-1
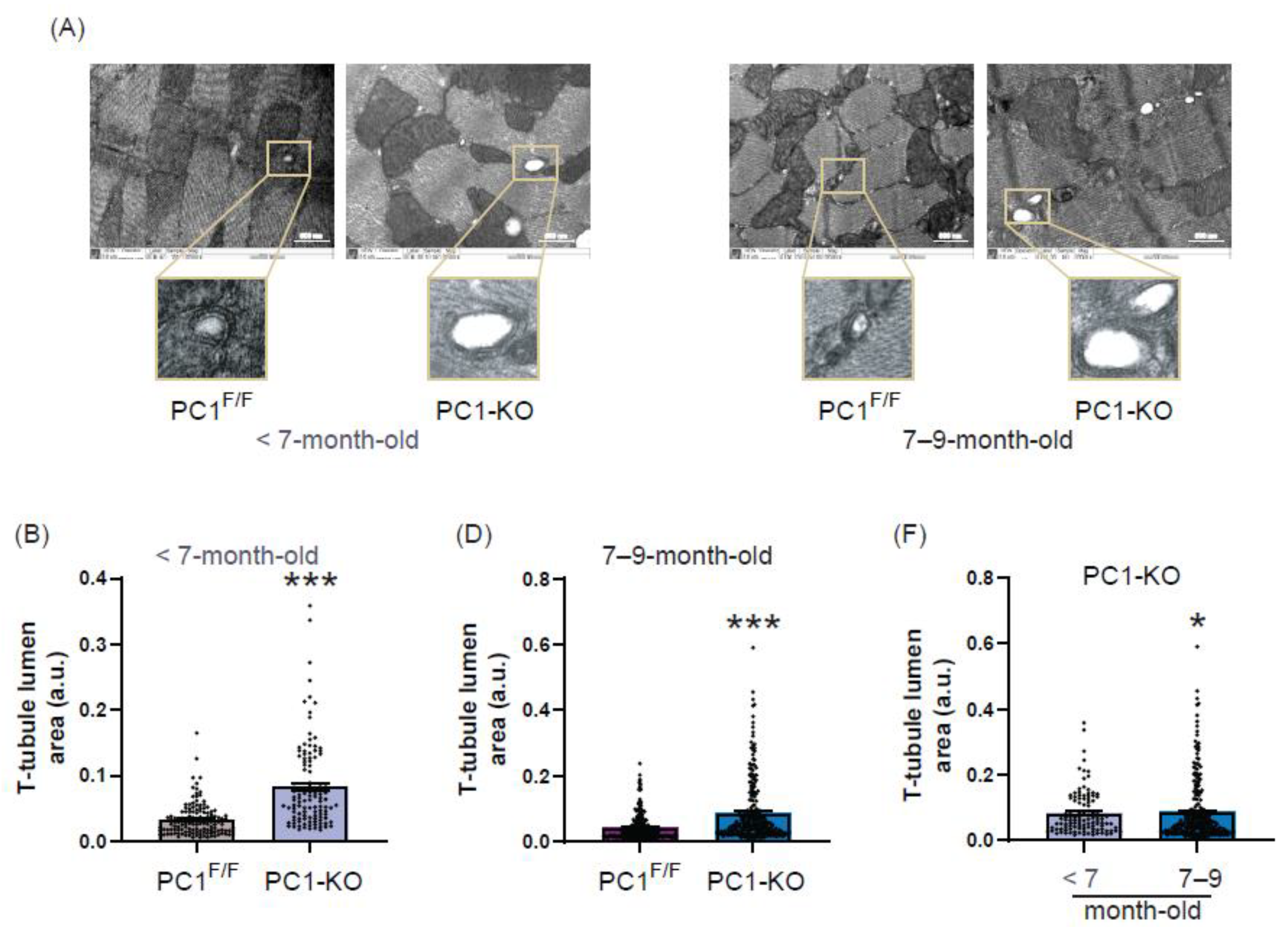
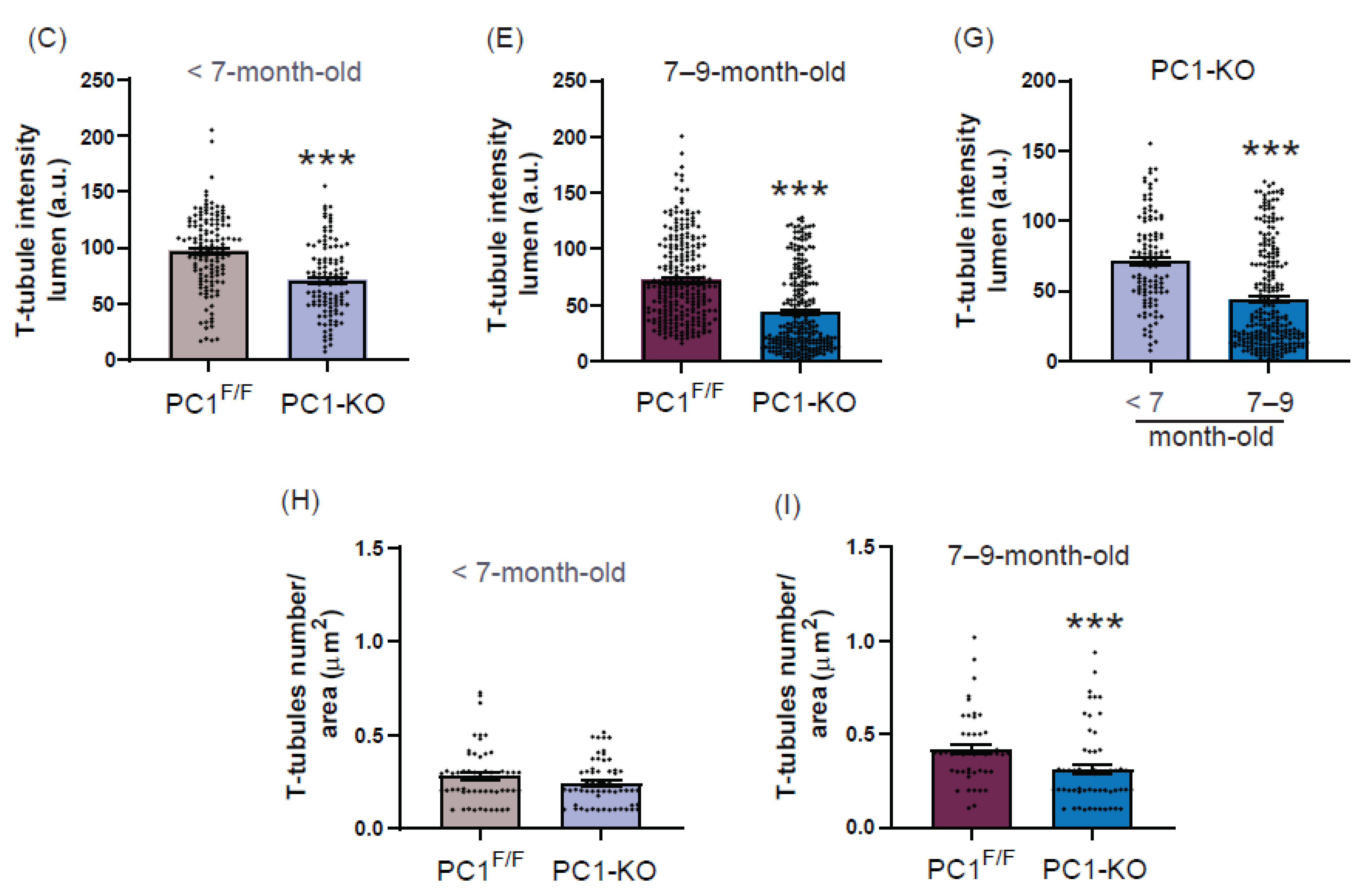
2.4. Ultrastructure of T-Tubules Correlates with Changes in Cardiomyocyte Polycystin-1 Expression
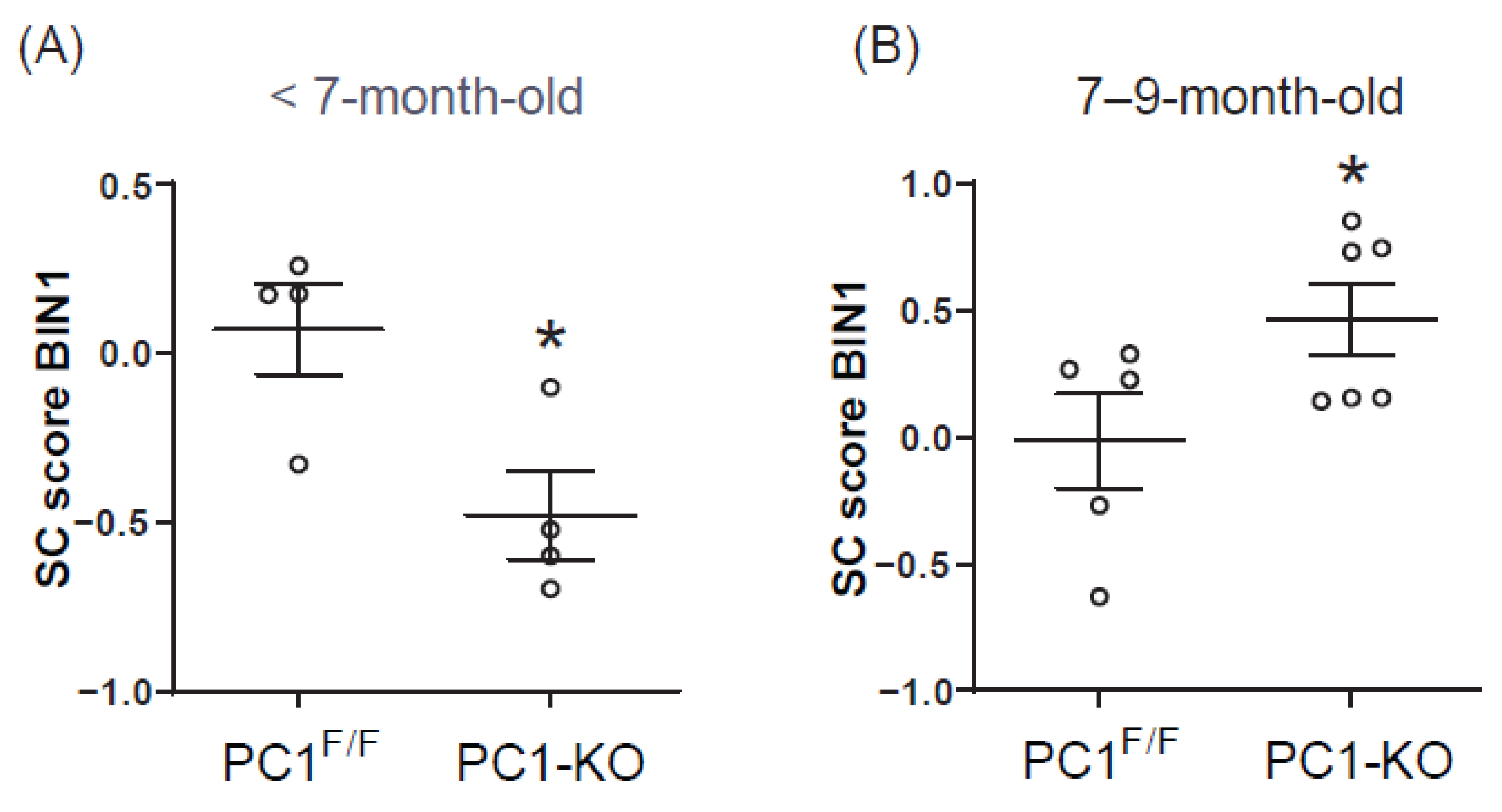
2.5. Plasma BIN1 + 13 + 17 Levels Correlate with Its Cardiac Expression in Cardiomyocytes Polycystin-1 Knockout Mice
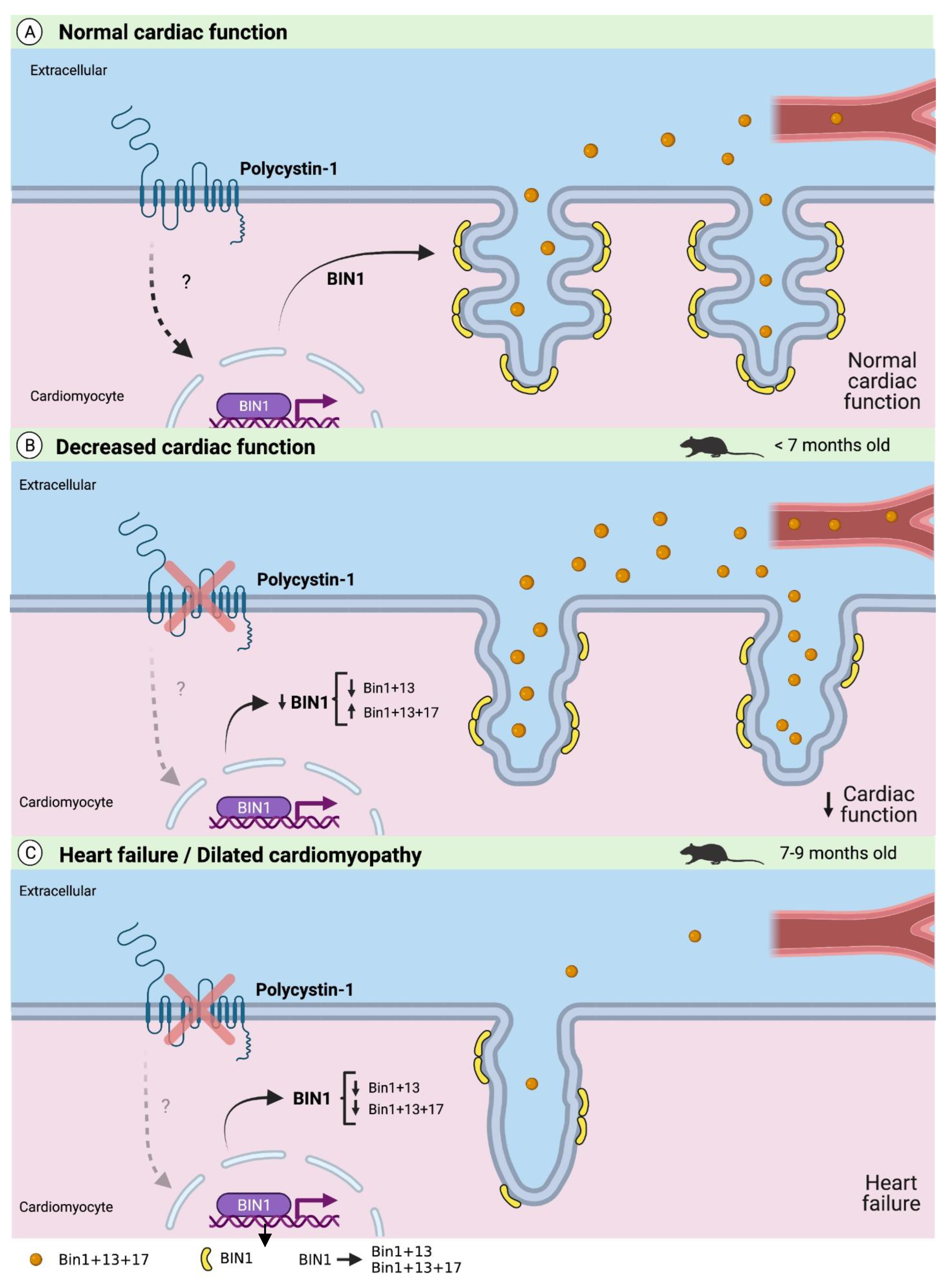
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Echocardiography and Morphometric Parameters
4.3. NRVM Culture and Transfection
4.4. Protein Extraction and Western Blot Analysis
4.5. Enrichment of Membrane Fraction
4.6. RNA Isolation and qRT-PCR
4.7. Histology
4.8. Transmission Electron Microscopy (TEM)
4.9. Plasma Cardiac BIN Content
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Delmas, P.; Padilla, F.; Osorio, N.; Coste, B.; Raoux, M.; Crest, M. Polycystins, calcium signaling, and human diseases. Biochem. Biophys. Res. Commun. 2004, 322, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Sutters, M. The Pathogenesis of Autosomal Dominant Polycystic Kidney Disease. Nephron-Exp. Nephrol. 2006, 103, e149–e155. [Google Scholar] [CrossRef] [PubMed]
- Ecder, T.; Schrier, R.W. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat. Rev. Nephrol. 2009, 5, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Hogan, M.C.; El-Zoghby, Z.M.; Irazabal, M.V.; Senum, S.R.; Heyer, C.M.; Madsen, C.D.; Cornec-Le Gall, E.; Behfar, A.; Harris, P.C.; et al. Autosomal Dominant Polycystic Kidney Patients May Be Predisposed to Various Cardiomyopathies. Kidney Int. Rep. 2017, 2, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Miura, A.; Kondo, H.; Yamamoto, T.; Okumura, Y.; Nishio, H. Sudden Unexpected Death of Infantile Dilated Cardiomyopathy with JPH2 and PKD1 Gene Variants. Int. Heart J. 2020, 61, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, D.A.L.; Kumanan, T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: A rare genetic asso-ciation. J. Ceylon Coll. Physicians 2016, 46, 42. [Google Scholar] [CrossRef]
- Malekshahabi, T.; Rad, N.K.; Serra, A.L.; Moghadasali, R. Autosomal dominant polycystic kidney disease: Disrupted pathways and potential therapeutic interventions. J. Cell. Physiol. 2019, 234, 12451–12470. [Google Scholar] [CrossRef] [PubMed]
- Pedrozo, Z.; Criollo, A.; Battiprolu, P.K.; Morales, C.R.; Contreras-Ferrat, A.; Fernández, C.; Jiang, N.; Luo, X.; Caplan, M.J.; Somlo, S.; et al. Polycystin-1 Is a Cardiomyocyte Mechanosensor That Governs L-Type Ca2+ Channel Protein Stability. Circulation 2015, 131, 2131–2142. [Google Scholar] [CrossRef]
- Altamirano, F.; Schiattarella, G.G.; French, K.M.; Kim, S.Y.; Engelberger, F.; Kyrychenko, S.; Villalobos, E.; Tong, D.; Schneider, J.W.; Ramirez-Sarmiento, C.A.; et al. Polycystin-1 Assembles With Kv Channels to Govern Cardiomyocyte Repolarization and Contractility. Circulation 2019, 140, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Yang, H.; Zhang, S.-S.; Cho, H.C.; Kalashnikova, M.; Sun, B.; Zhang, H.; Bhargava, A.; Grabe, M.; Olgin, J.; et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat. Med. 2014, 20, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hong, T.T. BIN1 regulates dynamic t-tubule membrane. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1839–1847. [Google Scholar] [CrossRef]
- Zhou, K.; Hong, T. Cardiac BIN1 (cBIN1) is a regulator of cardiac contractile function and an emerging biomarker of heart muscle health. Sci. China Life Sci. 2017, 60, 257–263. [Google Scholar] [CrossRef]
- Louch, W.E.; Trafford, A.W. Cardiac Transverse Tubules in Physiology and Heart Failure. Annu. Rev. Physiol. 2022, 84, 229–255. [Google Scholar] [CrossRef]
- Frost, A.; Unger, V.M.; De Camilli, P. The BAR Domain Superfamily: Membrane-Molding Macromolecules. Cell 2009, 137, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.-T.; Smyth, J.W.; Chu, K.Y.; Vogan, J.M.; Fong, T.S.; Jensen, B.C.; Fang, K.; Halushka, M.K.; Russell, S.D.; Colecraft, H.; et al. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm 2012, 9, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Laury-Kleintop, L.D.; Mulgrew, J.R.; Heletz, I.; Nedelcoviciu, R.A.; Chang, M.Y.; Harris, D.M.; Koch, W.J.; Schneider, M.D.; Muller, A.J.; Prendergast, G.C. Cardiac-Specific Disruption of Bin1 in Mice Enables a Model of Stress- and Age-Associated Dilated Cardio-myopathy. J. Cell. Biochem. 2015, 116, 2541–2551. [Google Scholar] [CrossRef]
- Caldwell, J.; Smith, C.; Taylor, R.; Kitmitto, A.; Eisner, D.; Dibb, K.; Trafford, A.W. Dependence of Cardiac Transverse Tubules on the BAR Domain Protein Amphiphysin II (BIN-1). Circ. Res. 2014, 115, 986–996. [Google Scholar] [CrossRef]
- Wei, S.; Guo, A.; Chen, B.; Kutschke, W.J.; Xie, Y.-P.; Zimmerman, K.; Weiss, R.M.; Anderson, M.E.; Cheng, H.; Song, L.-S. T-Tubule Remodeling During Transition From Hypertrophy to Heart Failure. Circ. Res. 2010, 107, 520–531. [Google Scholar] [CrossRef]
- Li, J.; Richmond, B.; Hong, T. Cardiac T-Tubule cBIN1-Microdomain, a Diagnostic Marker and Therapeutic Target of Heart Failure. Int. J. Mol. Sci. 2021, 22, 2299. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, A.P.; Hitzeman, T.C.; Baum, R.; Caldaruse, A.-M.; Agvanian, S.; Xie, Y.; Geft, D.R.; Chang, D.H.; Moriguchi, J.D.; Hage, A.; et al. Association of a Novel Diagnostic Biomarker, the Plasma Cardiac Bridging Integrator 1 Score, With Heart Failure With Preserved Ejection Fraction and Cardiovascular Hospitalization. JAMA Cardiol. 2018, 3, 1206–1210. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.-T.; Cogswell, R.; James, C.A.; Kang, G.; Pullinger, C.R.; Malloy, M.J.; Kane, J.P.; Wojciak, J.; Calkins, H.; Scheinman, M.M.; et al. Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2012, 9, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Perrone, R.D.; Ruthazer, R.; Terrin, N.C. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: Contribution of extrarenal complications to mortality. Am. J. Kidney Dis. 2001, 38, 777–784. [Google Scholar] [CrossRef]
- Lee, J.-J.; Cheng, S.-J.; Huang, C.-Y.; Chen, C.-Y.; Feng, L.; Hwang, D.-Y.; Kamp, T.J.; Chen, H.-C.; Hsieh, P.C. Primary cardiac manifestation of autosomal dominant polycystic kidney disease revealed by patient induced pluripotent stem cell-derived cardiomyocytes. Ebiomedicine 2019, 40, 675–684. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Chapman, A.B. Polycystins, ADPKD, and Cardiovascular Disease. Kidney Int. Rep. 2020, 5, 396–406. [Google Scholar] [CrossRef]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef]
- Schrier, R.W.; Johnson, A.M.; Mcfann, K.; Chapman, A.B. The role of parental hypertension in the frequency and age of diagnosis of hypertension in offspring with autosomal-dominant polycystic kidney disease. Kidney Int. 2003, 64, 1792–1799. [Google Scholar] [CrossRef][Green Version]
- Lyon, A.R.; MacLeod, K.T.; Zhang, Y.; Garcia, E.; Kanda, G.K.; Lab, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA 2009, 106, 6854–6859. [Google Scholar] [CrossRef]
- Yamakawa, S.; Wu, D.; Dasgupta, M.; Pedamallu, H.; Gupta, B.; Modi, R.; Mufti, M.; O’Callaghan, C.; Frisk, M.; Louch, W.E.; et al. Role of t-tubule remodeling on mechanisms of abnormal calcium release during heart failure development in canine ventricle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1658–H1669. [Google Scholar] [CrossRef]
- Lipsett, D.B.; Frisk, M.; Aronsen, J.M.; Nordén, E.S.; Buonarati, O.R.; Cataliotti, A.; Hell, J.W.; Sjaastad, I.; Christensen, G.; Louch, W.E. Cardiomyocyte substructure reverts to an immature phenotype during heart failure. J. Physiol. 2019, 597, 1833–1853. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.; Ruud, M.; Espe, E.K.S.; Aronsen, J.M.; Røe, T.; Zhang, L.; Norseng, P.A.; Sejersted, O.M.; Christensen, G.A.; Sjaastad, I.; et al. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 2016, 112, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Setterberg, I.E.; Le, C.; Frisk, M.; Li, J.; Louch, W.E. The Physiology and Pathophysiology of T-Tubules in the Heart. Front. Physiol. 2021, 12, 1–21. [Google Scholar] [CrossRef]
- Hong, T.; Shaw, R.M. Cardiac T-Tubule Microanatomy and Function. Physiol. Rev. 2017, 97, 227–252. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, K.; Li, J.; Agvanian, S.; Caldaruse, A.-M.; Shaw, S.; Hitzeman, T.C.; Shaw, R.M.; Hong, T. In Mice Subjected to Chronic Stress, Exogenous cBIN1 Preserves Calcium-Handling Machinery and Cardiac Function. JACC Basic Transl. Sci. 2020, 5, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tian, Q.; Barth, M.; Xian, W.; Ruppenthal, S.; Schaefers, H.-J.; Chen, Z.; Moretti, A.; Laugwitz, K.-L.; Lipp, P. Human BIN1 isoforms grow, maintain, and regenerate excitation–contraction couplons in adult rat and human stem cell-derived cardiomyocytes. Cardiovasc. Res. 2022, 118, 1479–1491. [Google Scholar] [CrossRef]
- Li, J.; Agvanian, S.; Zhou, K.; Shaw, R.M.; Hong, T. Exogenous Cardiac Bridging Integrator 1 Benefits Mouse Hearts With Pre-existing Pressure Overload-Induced Heart Failure. Front. Physiol. 2020, 11, 708. [Google Scholar] [CrossRef]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. c-MYC Suppresses BIN1 to Release Poly(ADP-Ribose) Polymerase 1: A Mechanism by Which Cancer Cells Acquire Cisplatin Resistance. Sci. Signal. 2011, 4, ra19. [Google Scholar] [CrossRef]
- Lee, E.J.; Seo, E.; Kim, J.W.; Nam, S.A.; Lee, J.Y.; Jun, J.; Oh, S.; Park, M.; Jho, E.-H.; Yoo, K.H.; et al. TAZ/Wnt-β-catenin/c-MYC axis regulates cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2020, 117, 29001–29012. [Google Scholar] [CrossRef]
- Córdova-Casanova, A.; Olmedo, I.; Riquelme, J.A.; Barrientos, G.; Sánchez, G.; Gillette, T.G.; Lavandero, S.; Chiong, M.; Donoso, P.; Pedrozo, Z. Mechanical stretch increases L-type calcium channel stability in cardiomyocytes through a polycys-tin-1/AKT-dependent mechanism. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 289–296. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Vesga, M.C.; Flores-Vergara, R.; Riquelme, J.A.; Llancaqueo, M.; Sánchez, G.; Vergara, C.; Michea, L.; Donoso, P.; Quest, A.F.G.; Olmedo, I.; et al. Polycystin-1 Is a Crucial Regulator of BIN1 Expression and T-Tubule Remodeling Associated with the Development of Dilated Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 667. https://doi.org/10.3390/ijms24010667
Díaz-Vesga MC, Flores-Vergara R, Riquelme JA, Llancaqueo M, Sánchez G, Vergara C, Michea L, Donoso P, Quest AFG, Olmedo I, et al. Polycystin-1 Is a Crucial Regulator of BIN1 Expression and T-Tubule Remodeling Associated with the Development of Dilated Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(1):667. https://doi.org/10.3390/ijms24010667
Chicago/Turabian StyleDíaz-Vesga, Magda C., Raúl Flores-Vergara, Jaime A. Riquelme, Marcelo Llancaqueo, Gina Sánchez, Cecilia Vergara, Luis Michea, Paulina Donoso, Andrew F. G. Quest, Ivonne Olmedo, and et al. 2023. "Polycystin-1 Is a Crucial Regulator of BIN1 Expression and T-Tubule Remodeling Associated with the Development of Dilated Cardiomyopathy" International Journal of Molecular Sciences 24, no. 1: 667. https://doi.org/10.3390/ijms24010667
APA StyleDíaz-Vesga, M. C., Flores-Vergara, R., Riquelme, J. A., Llancaqueo, M., Sánchez, G., Vergara, C., Michea, L., Donoso, P., Quest, A. F. G., Olmedo, I., & Pedrozo, Z. (2023). Polycystin-1 Is a Crucial Regulator of BIN1 Expression and T-Tubule Remodeling Associated with the Development of Dilated Cardiomyopathy. International Journal of Molecular Sciences, 24(1), 667. https://doi.org/10.3390/ijms24010667