
Evidence for a Conserved Function of Eukaryotic Pantothenate Kinases in the Regulation of Mitochondrial Homeostasis and Oxidative Stress

 , , , , and
, , , , and 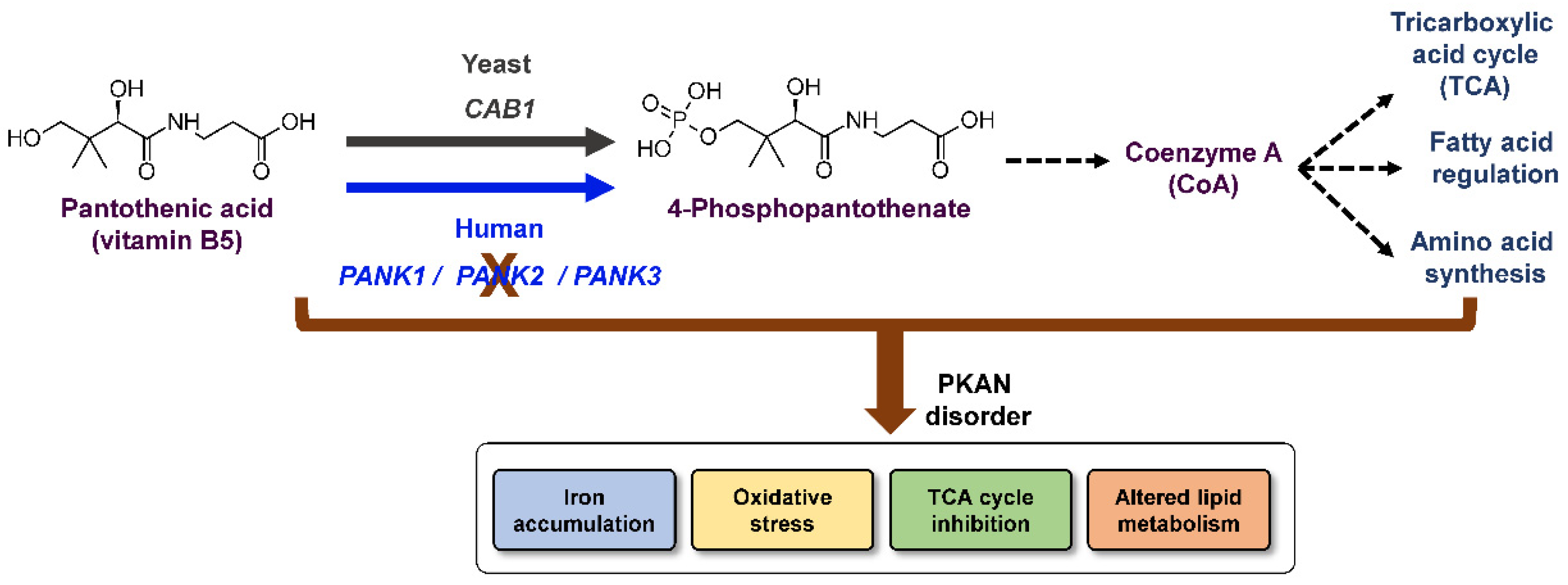{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
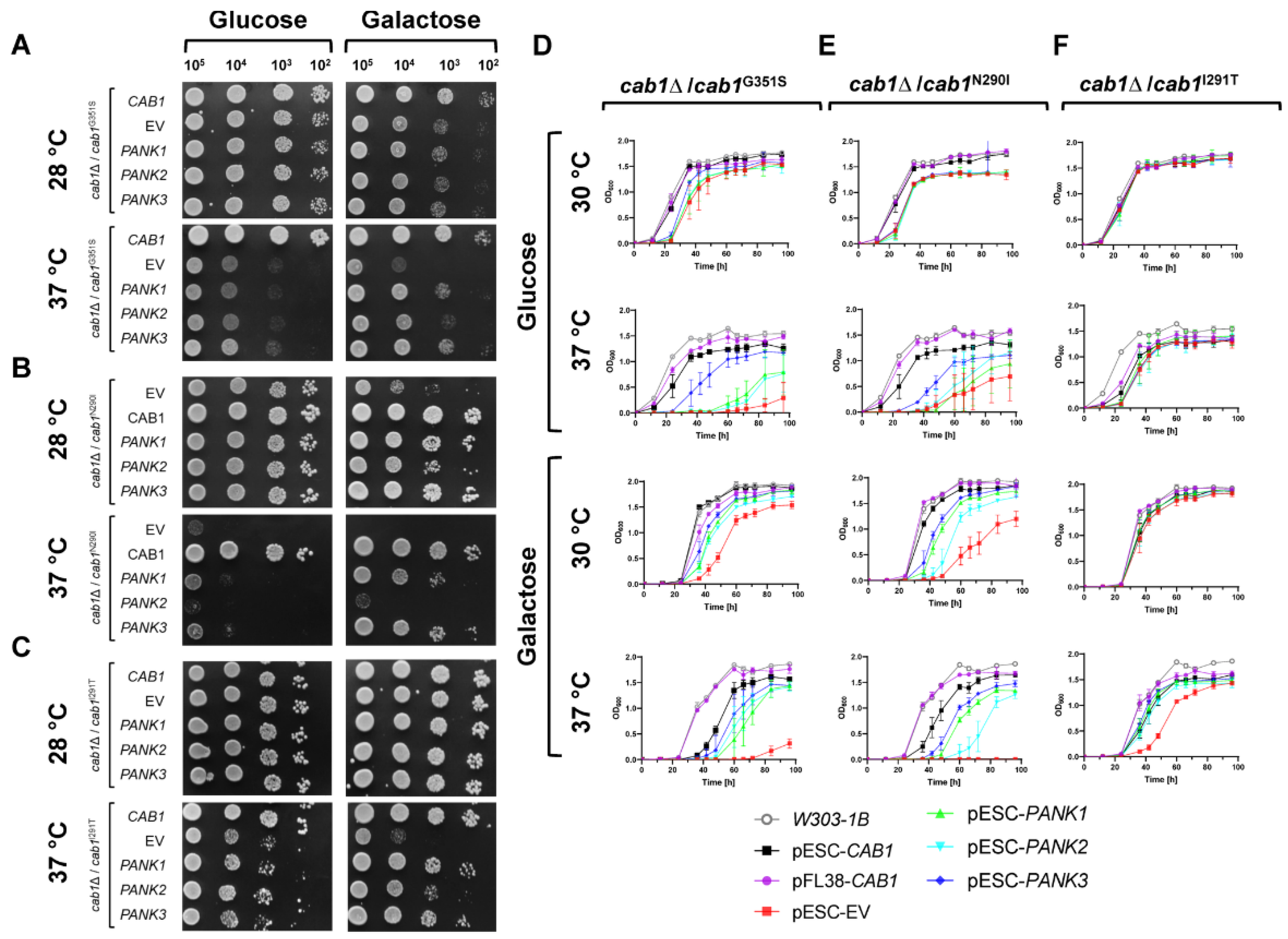
2.1. Functional Analysis of Human PANK1, PANK2, and PANK3 in Yeast
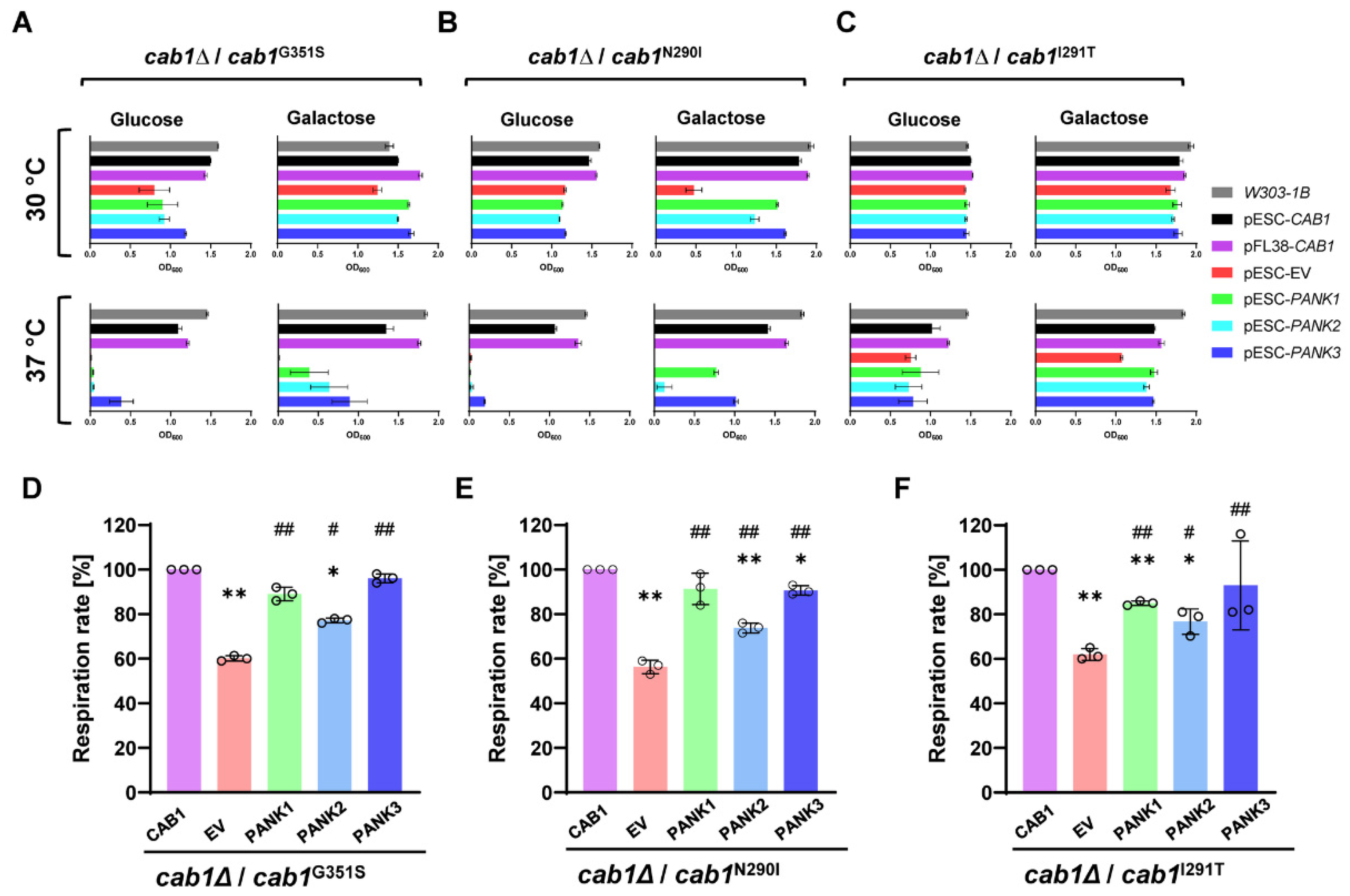
2.2. Complementation Analysis of Respiratory Activity Deficiency of cab1 Mutant Strains
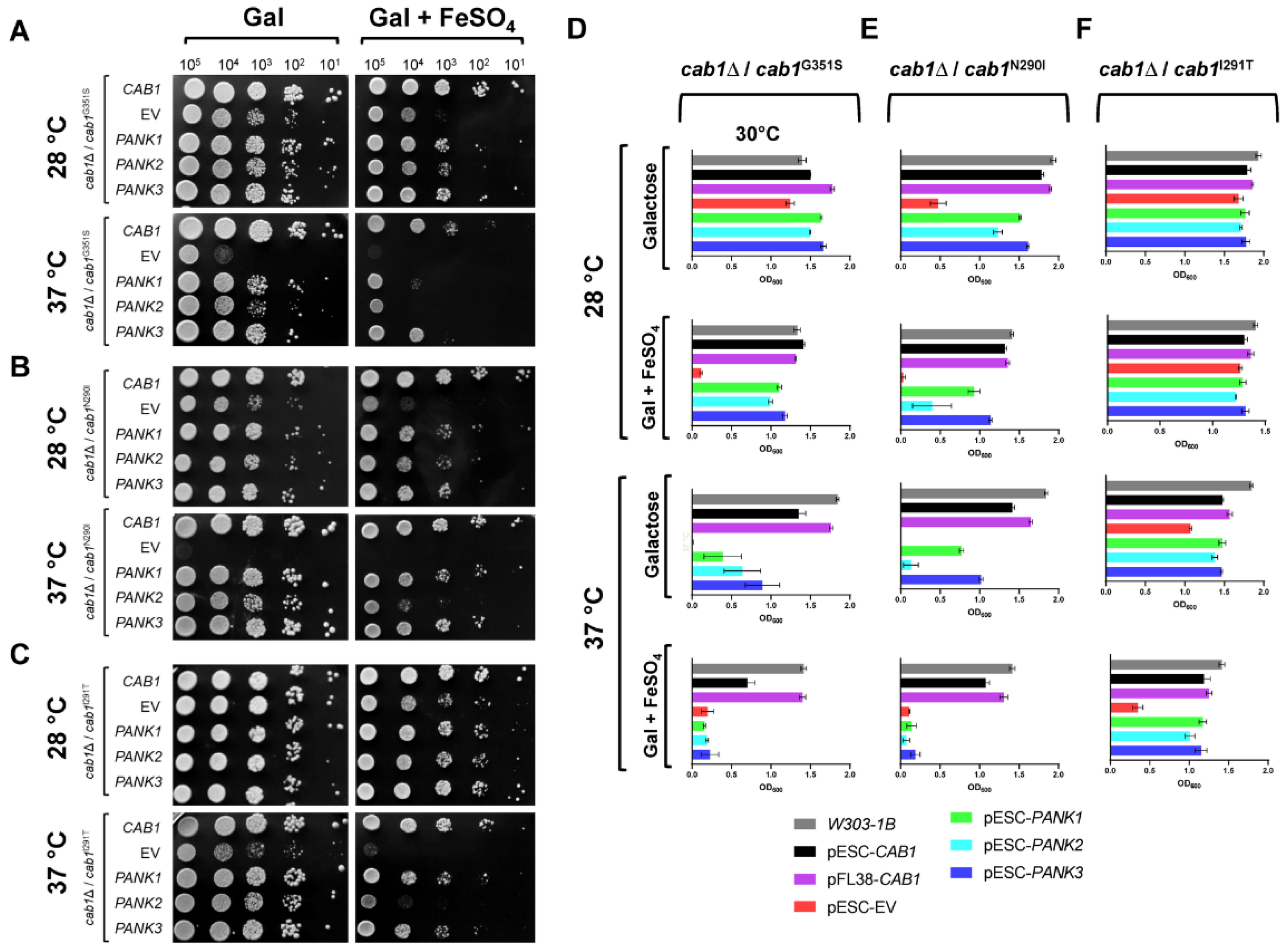
2.3. Human PanKs Complement Iron Susceptibility of Yeast cab1 Mutants
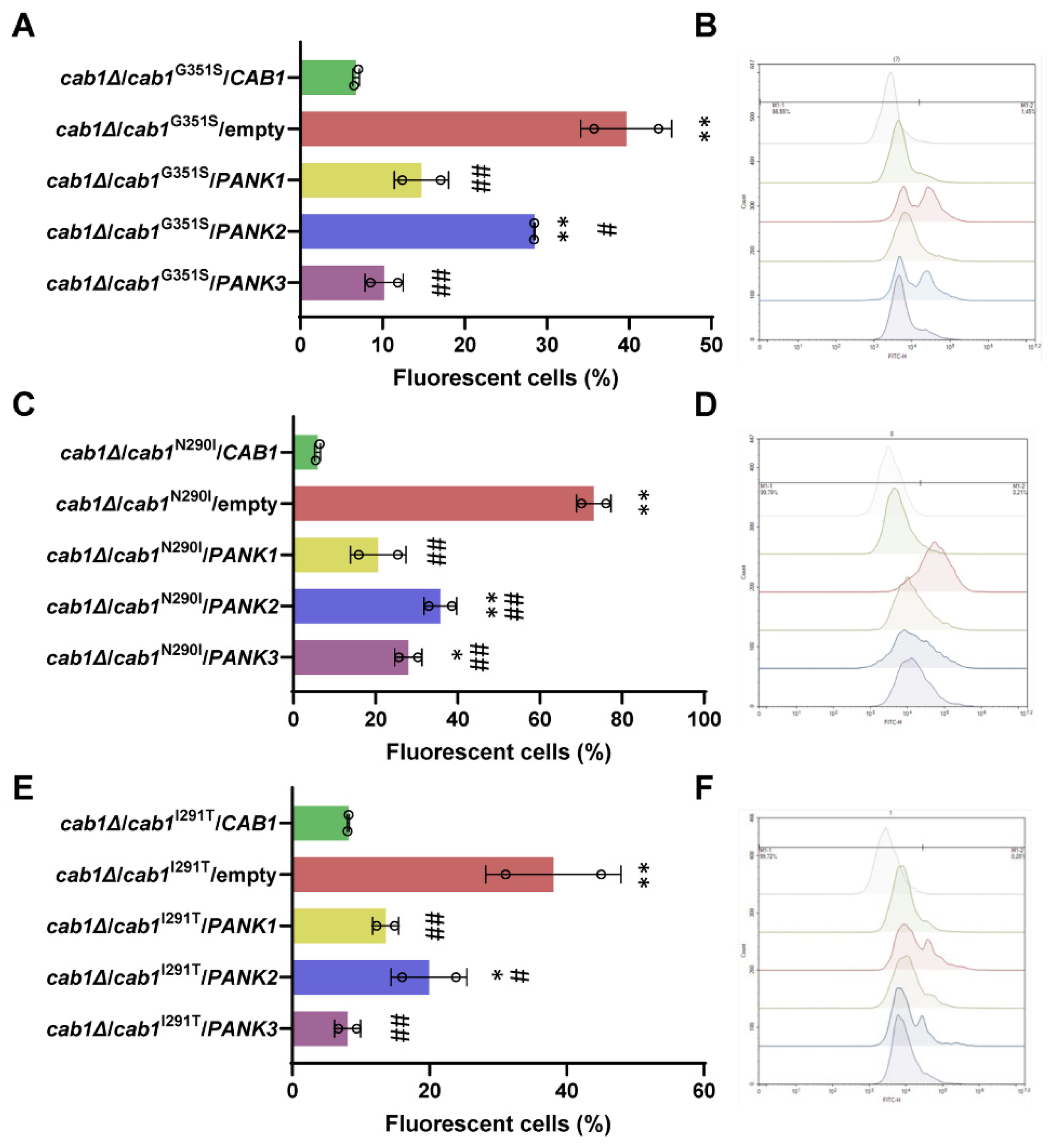
2.4. Complementation of the Oxidative Damage of CAB1 Defective Yeast Mutants by Human PanKs
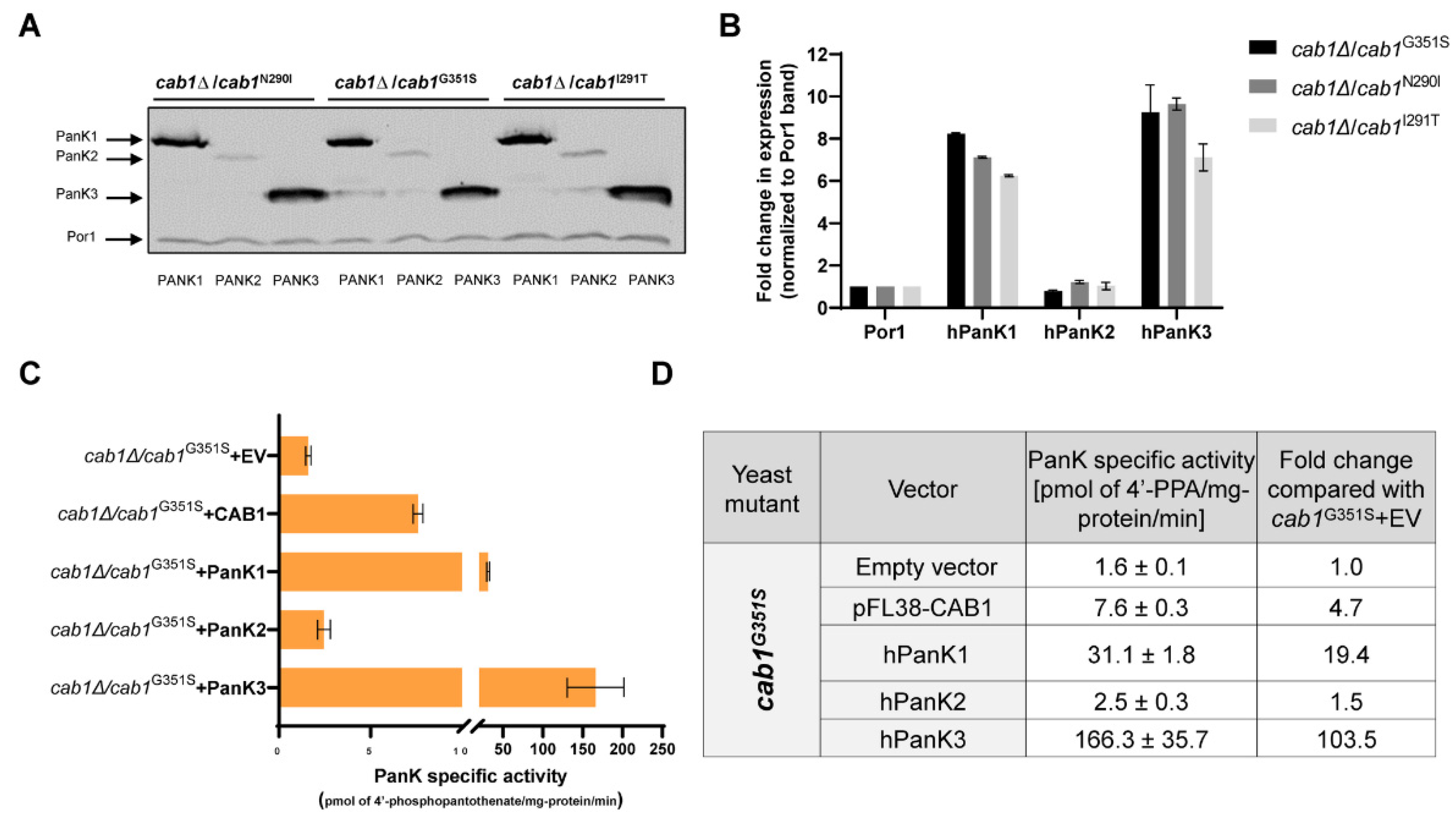
2.5. Correlation between Complementation Ability and Human PanK Expression and Activity
3. Discussion
4. Materials and Methods
4.1. Yeast Strains, Media, and Plasmid Construction
4.2. Yeast Complementation and Iron Sensitivity Spotting Assays
4.3. Oxygen Consumption Rate (OCR) and Reactive Oxygen Species (ROS) Measurements
4.4. Monitoring of hPanK Steady-State Expression in Yeast
4.5. Liquid Yeast Growth Assays
4.6. PanK Enzymatic Activity in Yeasts
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olzhausen, J.; Schübbe, S.; Schüller, H.-J. Genetic Analysis of Coenzyme A Biosynthesis in the Yeast Saccharomyces Cerevisiae: Identification of a Conditional Mutation in the Pantothenate Kinase Gene CAB1. Curr. Genet. 2009, 55, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Dansie, L.E.; Reeves, S.; Miller, K.; Zano, S.P.; Frank, M.; Pate, C.; Wang, J.; Jackowski, S. Physiological Roles of the Pantothenate Kinases. Biochem Soc Trans 2014, 42, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A, Protein CoAlation and Redox Regulation in Mammalian Cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Begley, T.P.; Kinsland, C.; Strauss, E. The Biosynthesis of Coenzyme A in Bacteria. Vitam. Horm. 2001, 61, 157–171. [Google Scholar] [CrossRef]
- Chiu, J.E.; Thekkiniath, J.; Choi, J.-Y.; Perrin, B.A.; Lawres, L.; Plummer, M.; Virji, A.Z.; Abraham, A.; Toh, J.Y.; Zandt, M.V.; et al. The Antimalarial Activity of the Pantothenamide α-PanAm Is via Inhibition of Pantothenate Phosphorylation. Sci. Rep. 2017, 7, 14234. [Google Scholar] [CrossRef]
- Huang, L.; Khusnutdinova, A.; Nocek, B.; Brown, G.; Xu, X.; Cui, H.; Petit, P.; Flick, R.; Zallot, R.; Balmant, K.; et al. A Family of Metal-Dependent Phosphatases Implicated in Metabolite Damage-Control. Nat. Chem. Biol. 2016, 12, 621–627. [Google Scholar] [CrossRef]
- Yao, J.; Subramanian, C.; Rock, C.O.; Jackowski, S. Human Pantothenate Kinase 4 Is a Pseudo-Pantothenate Kinase. Protein Sci. 2019, 28, 1031–1047. [Google Scholar] [CrossRef]
- Dibble, C.C.; Barritt, S.A.; Perry, G.E.; Lien, E.C.; Geck, R.C.; DuBois-Coyne, S.E.; Bartee, D.; Zengeya, T.T.; Cohen, E.B.; Yuan, M.; et al. PI3K Drives the de Novo Synthesis of Coenzyme A from Vitamin B5. Nature 2022, 608, 192–198. [Google Scholar] [CrossRef]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A Novel Pantothenate Kinase Gene (PANK2) Is Defective in Hallervorden-Spatz Syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef]
- Alfonso-Pecchio, A.; Garcia, M.; Leonardi, R.; Jackowski, S. Compartmentalization of Mammalian Pantothenate Kinases. PLoS One 2012, 7, e49509. [Google Scholar] [CrossRef]
- Johnson, M.A.; Kuo, Y.M.; Westaway, S.K.; Parker, S.M.; Ching, K.H.L.; Gitschier, J.; Hayflick, S.J. Mitochondrial Localization of Human PANK2 and Hypotheses of Secondary Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1012, 282–298. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Truax, A.C.; Trojanowski, J.Q.; Lee, V.M.-Y. Altered Neuronal Mitochondrial Coenzyme A Synthesis in Neurodegeneration with Brain Iron Accumulation Caused by Abnormal Processing, Stability, and Catalytic Activity of Mutant Pantothenate Kinase 2. J. Neurosci. 2005, 25, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Munshi, M.I.; Yao, S.J.; Ben Mamoun, C. Redesigning Therapies for Pantothenate Kinase-Associated Neurodegeneration. J. Biol. Chem. 2022, 298, 101577. [Google Scholar] [CrossRef] [PubMed]
- Hörtnagel, K.; Prokisch, H.; Meitinger, T. An Isoform of HPANK2, Deficient in Pantothenate Kinase-Associated Neurodegeneration, Localizes to Mitochondria. Hum. Mol. Genet. 2003, 12, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Hogarth, P.; Placzek, A.; Gregory, A.M.; Fox, R.; Zhen, D.; Hamada, J.; van der Zwaag, M.; Lambrechts, R.; Jin, H.; et al. 4′-Phosphopantetheine Corrects CoA, Iron, and Dopamine Metabolic Defects in Mammalian Models of PKAN. EMBO Mol. Med. 2019, 11, e10489. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Yao, J.; Frank, M.W.; Rock, C.O.; Jackowski, S. A Pantothenate Kinase-Deficient Mouse Model Reveals a Gene Expression Program Associated with Brain Coenzyme a Reduction. Biochim. Biophy.s Acta Mol. Basis Dis. 2020, 1866, 165663. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J. Neurodegeneration with Brain Iron Accumulation: From Genes to Pathogenesis. Semin. Pediatr. Neurol. 2006, 13, 182–185. [Google Scholar] [CrossRef]
- Gregory, A.; Hayflick, S.J. Neurodegeneration with Brain Iron Accumulation. Folia Neuropathol. 2005, 43, 286–296. [Google Scholar]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinós, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Masud, A.J.; Kastaniotis, A.J.; Rahman, M.T.; Autio, K.J.; Hiltunen, J.K. Mitochondrial Acyl Carrier Protein (ACP) at the Interface of Metabolic State Sensing and Mitochondrial Function. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 118540. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Li, C.; Lv, S.; Zhou, B. Pantothenate Kinase-Associated Neurodegeneration: Insights from a Drosophila Model. Hum Mol. Genet. 2009, 18, 3659–3672. [Google Scholar] [CrossRef]
- Gihaz, S.; Gareiss, P.; Choi, J.-Y.; Renard, I.; Pal, A.C.; Surovsteva, Y.; Chiu, J.E.; Thekkiniath, J.; Plummer, M.; Hungerford, W.; et al. High-Resolution Crystal Structure and Chemical Screening Reveal Pantothenate Kinase as a New Target for Antifungal Development. Structure 2022, 30, 1494–1507.e6. [Google Scholar] [CrossRef] [PubMed]
- Ceccatelli Berti, C.; Gilea, A.I.; De Gregorio, M.A.; Goffrini, P. Exploring Yeast as a Study Model of Pantothenate Kinase-Associated Neurodegeneration and for the Identification of Therapeutic Compounds. Int. J. Mol. Sci. 2020, 22, 293. [Google Scholar] [CrossRef]
- Chiu, J.E.; Thekkiniath, J.; Mehta, S.; Müller, C.; Bracher, F.; Ben Mamoun, C. The Yeast Pantothenate Kinase Cab1 Is a Master Regulator of Sterol Metabolism and of Susceptibility to Ergosterol Biosynthesis Inhibitors. J. Biol. Chem. 2019, 294, 14757–14767. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Cazzalini, O. Deletion of the Yeast Homologue of the Human Gene Associated with Friedreich’s Ataxia Elicits Iron Accumulation in Mitochondria. FEBS Lett. 1997, 411, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Finazzi, D. Neurodegeneration with Brain Iron Accumulation: Update on Pathogenic Mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Satoh, A.; Horinouchi, Y.; Hamano, H.; Watanabe, H.; Imao, M.; Imanishi, M.; Zamami, Y.; Takechi, K.; Izawa-Ishizawa, Y.; et al. Iron Accumulation Causes Impaired Myogenesis Correlated with MAPK Signaling Pathway Inhibition by Oxidative Stress. FASEB J. 2019, 33, 9551–9564. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Núñez, M.T. The Interplay between Iron Accumulation, Mitochondrial Dysfunction, and Inflammation during the Execution Step of Neurodegenerative Disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.-J.; Kayser, O.; Sibon, O.C.M. Pantethine Rescues a Drosophila Model for Pantothenate Kinase-Associated Neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef]
- Srinivasan, B.; Baratashvili, M.; van der Zwaag, M.; Kanon, B.; Colombelli, C.; Lambrechts, R.A.; Schaap, O.; Nollen, E.A.; Podgoršek, A.; Kosec, G.; et al. Extracellular 4′-Phosphopantetheine Is a Source for Intracellular Coenzyme A Synthesis. Nat. Chem. Biol. 2015, 11, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. IPSC-Derived Neuronal Models of PANK2-Associated Neurodegeneration Reveal Mitochondrial Dysfunction Contributing to Early Disease. PLoS One 2017, 12, e0184104. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Grzeschik, N.A.; et al. CoA-Dependent Activation of Mitochondrial Acyl Carrier Protein Links Four Neurodegenerative Diseases. EMBO Mol. Med. 2019, 11, e10488. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.-K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global Analysis of Protein Localization in Budding Yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Subramanian, C.; Yun, M.-K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A Therapeutic Approach to Pantothenate Kinase Associated Neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef]
- Thakur, N.; Klopstock, T.; Jackowski, S.; Kuscer, E.; Tricta, F.; Videnovic, A.; Jinnah, H.A. Rational Design of Novel Therapies for Pantothenate Kinase-Associated Neurodegeneration. Mov. Disord. 2021, 36, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A Corrects Pathological Defects in Human Neurons of PANK2-Associated Neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef]
- Gietz, R.D. Yeast Transformation by the LiAc/SS Carrier DNA/PEG Method. Methods Mol. Biol. 2014, 1205, 1–12. [Google Scholar] [CrossRef]
- Goffrini, P.; Ercolino, T.; Panizza, E.; Giachè, V.; Cavone, L.; Chiarugi, A.; Dima, V.; Ferrero, I.; Mannelli, M. Functional Study in a Yeast Model of a Novel Succinate Dehydrogenase Subunit B Gene Germline Missense Mutation (C191Y) Diagnosed in a Patient Affected by a Glomus Tumor. Hum. Mol. Genet. 2009, 18, 1860–1868. [Google Scholar] [CrossRef]
- di Punzio, G.; Di Noia, M.A.; Delahodde, A.; Sellem, C.; Donnini, C.; Palmieri, L.; Lodi, T.; Dallabona, C. A Yeast-Based Screening Unravels Potential Therapeutic Molecules for Mitochondrial Diseases Associated with Dominant ANT1 Mutations. Int. J. Mol. Sci. 2021, 22, 4461. [Google Scholar] [CrossRef]
- Gomez, M.; Pérez-Gallardo, R.V.; Sánchez, L.A.; Díaz-Pérez, A.L.; Cortés-Rojo, C.; Meza Carmen, V.; Saavedra-Molina, A.; Lara-Romero, J.; Jiménez-Sandoval, S.; Rodríguez, F.; et al. Malfunctioning of the Iron-Sulfur Cluster Assembly Machinery in Saccharomyces Cerevisiae Produces Oxidative Stress via an Iron-Dependent Mechanism, Causing Dysfunction in Respiratory Complexes. PLoS One 2014, 9, e111585. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccatelli Berti, C.; Gihaz, S.; Figuccia, S.; Choi, J.-Y.; Pal, A.C.; Goffrini, P.; Ben Mamoun, C. Evidence for a Conserved Function of Eukaryotic Pantothenate Kinases in the Regulation of Mitochondrial Homeostasis and Oxidative Stress. Int. J. Mol. Sci. 2023, 24, 435. https://doi.org/10.3390/ijms24010435
Ceccatelli Berti C, Gihaz S, Figuccia S, Choi J-Y, Pal AC, Goffrini P, Ben Mamoun C. Evidence for a Conserved Function of Eukaryotic Pantothenate Kinases in the Regulation of Mitochondrial Homeostasis and Oxidative Stress. International Journal of Molecular Sciences. 2023; 24(1):435. https://doi.org/10.3390/ijms24010435
Chicago/Turabian StyleCeccatelli Berti, Camilla, Shalev Gihaz, Sonia Figuccia, Jae-Yeon Choi, Anasuya C. Pal, Paola Goffrini, and Choukri Ben Mamoun. 2023. "Evidence for a Conserved Function of Eukaryotic Pantothenate Kinases in the Regulation of Mitochondrial Homeostasis and Oxidative Stress" International Journal of Molecular Sciences 24, no. 1: 435. https://doi.org/10.3390/ijms24010435
APA StyleCeccatelli Berti, C., Gihaz, S., Figuccia, S., Choi, J.-Y., Pal, A. C., Goffrini, P., & Ben Mamoun, C. (2023). Evidence for a Conserved Function of Eukaryotic Pantothenate Kinases in the Regulation of Mitochondrial Homeostasis and Oxidative Stress. International Journal of Molecular Sciences, 24(1), 435. https://doi.org/10.3390/ijms24010435