
Expanding the Etiology of Oculo–Auriculo–Vertebral Spectrum: A Novel Interstitial Microdeletion at 1p36


Abstract
1. Introduction
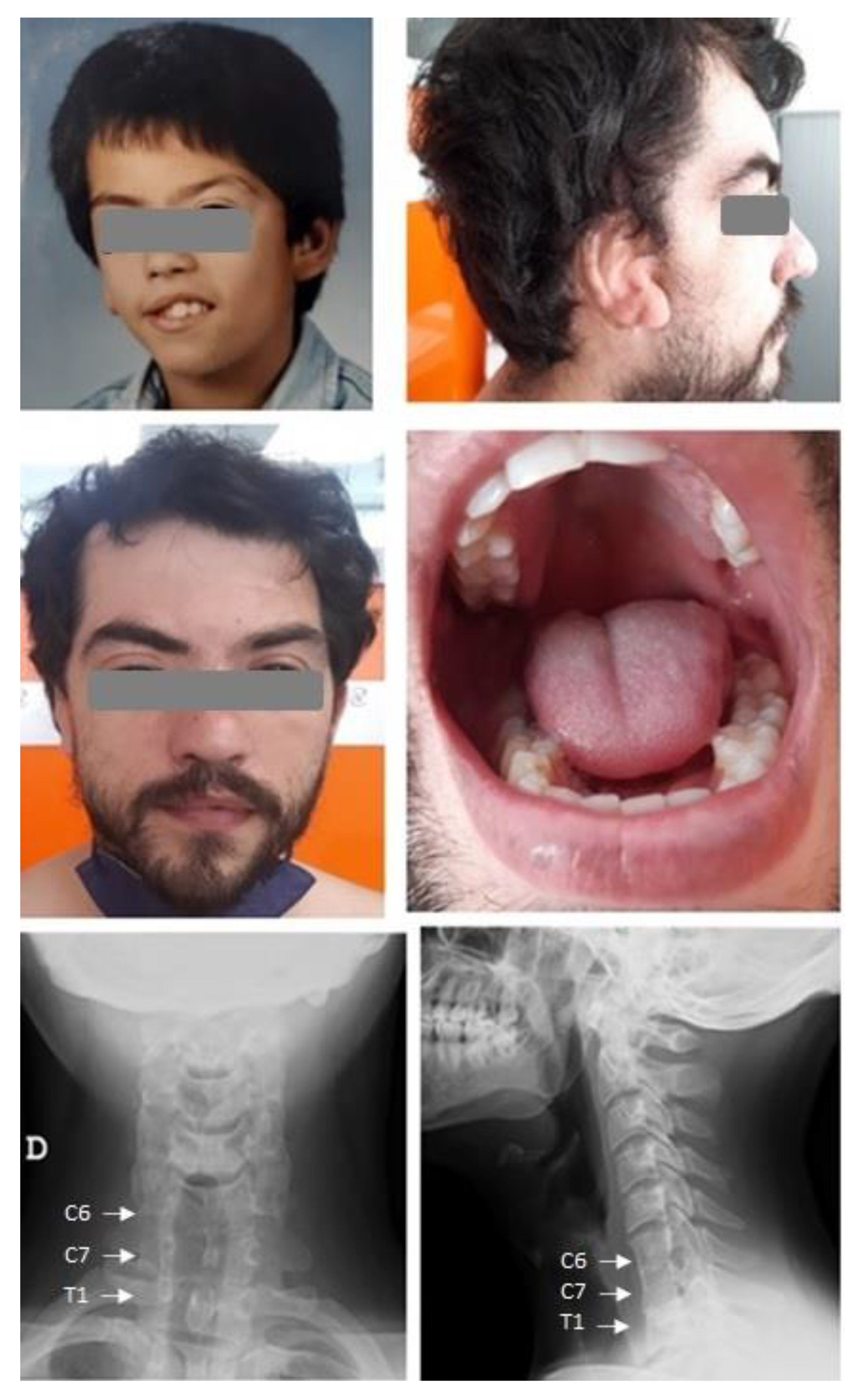
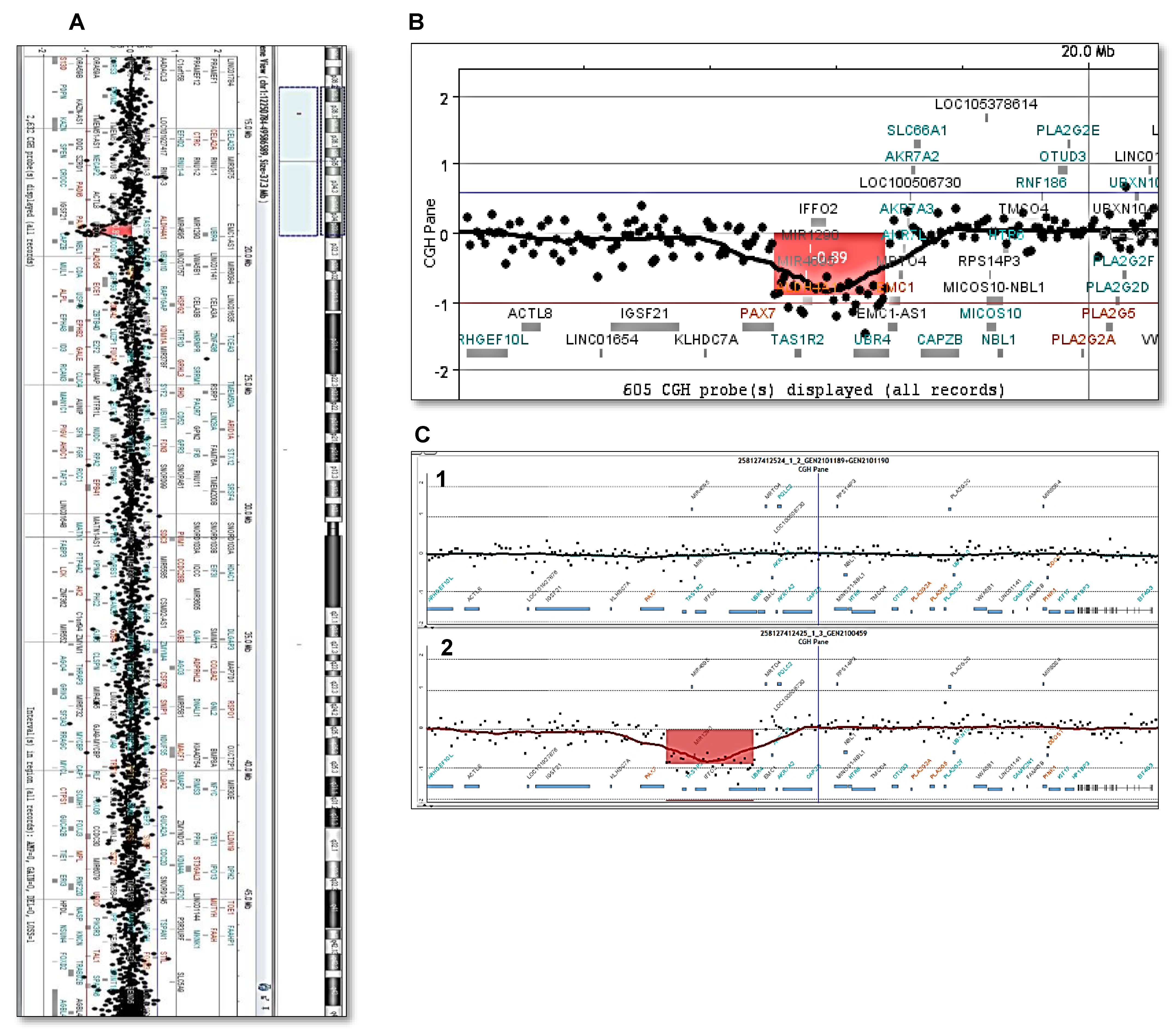
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beleza-Meireles, A.; Hart, R.; Clayton-Smith, J.; Oliveira, R.; Reis, C.F.; Venâncio, M.; Ramos, F.; Sá, J.; Ramos, L.; Cunha, E.; et al. Oculo-auriculo-vertebral spectrum: Clinical and molecular analysis of 51 patients. Eur. J. Med. Genet. 2015, 58, 455–465. [Google Scholar] [CrossRef]
- Cohen, N.; Cohen, E.; Gaiero, A.; Zecca, S.; Fichera, G.; Baldi, F.; Giordanetto, J.F.; Mercier, J.M.; Cohen, A. Maxillofacial features and systemic malformations in expanded spectrum Hemifacial Microsomia. Am. J. Med. Genet. Part A 2017, 173, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Bartel-Friedrich, S. Congenital Auricular Malformations: Description of Anomalies and Syndromes. Facial Plast. Surg. 2015, 31, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Gendron, C.; Schwentker, A.; van Aalst, J. Genetic Advances in the Understanding of Microtia. J. Pediatr. Genet. 2016, 5, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Tingaud-Sequeira, A.; Trimouille, A.; Sagardoy, T.; Lacombe, D.; Rooryck, C. Oculo-auriculo-vertebral spectrum: New genes and literature review on a complex disease. J. Med. Genet. 2022, 59, 417–427. [Google Scholar] [CrossRef]
- Estandia-Ortega, B.; Fernández-Hernández, L.; Alcántara-Ortigoza, M.A.; González-del Angel, A. Proposed clinical approach and imaging studies in families with oculo-auriculo-vertebral spectrum to assess variable expressivity. Am. J. Med. Genet. Part A 2022, 188, 1515–1525. [Google Scholar] [CrossRef]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. Part C 2017, 175, 70–115. [Google Scholar] [CrossRef]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef]
- Vishwanath, N.; Monis, W.J.; Hoffmann, G.A.; Ramachandran, B.; DiGiacomo, V.; Wong, J.Y.; Smith, M.L.; Layne, M.D. Mechanisms of aortic carboxypeptidase-like protein secretion and identification of an intracellularly retained variant associated with Ehlers-Danlos syndrome. J. Biol. Chem. 2020, 295, 9725–9735. [Google Scholar] [CrossRef]
- Tingaud-Sequeira, A.; Trimouille, A.; Salaria, M.; Stapleton, R.; Claverol, S.; Plaisant, C.; Bonneu, M.; Lopez, E.; Arveiler, B.; Lacombe, D.; et al. A recurrent missense variant in EYA3 gene is associated with oculo-auriculo-vertebral spectrum. Hum. Genet. 2021, 140, 933–944. [Google Scholar] [CrossRef]
- Carter, S.; Fellows, B.J.; Gibson, K.; Bicknell, L.S. Extending the PAX1 spectrum: A dominantly inherited variant causes oculo-auriculo-vertebral syndrome. Eur. J. Hum. Genet. 2022, 30, 1178–1181. [Google Scholar] [CrossRef]
- Guida, V.; Sparascio, F.P.; Bernardini, L.; Pancheri, F.; Melis, D.; Cocciadiferro, D.; Pagnoni, M.; Puzzo, M.; Goldoni, M.; Barone, C.; et al. Copy number variation analysis implicates novel pathways in patients with oculo-auriculo-vertebral-spectrum and congenital heart defects. Clin. Genet. 2021, 100, 268–279. [Google Scholar] [CrossRef]
- Callier, P.; Faivre, L.; Thauvin-Robinet, C.; Marle, N.; Mosca, A.L.; D’Athis, P.; Guy, J.; Masurel-Paulet, A.; Joly, L.; Guiraud, S.; et al. Array-CGH in a series of 30 patients with mental retardation, dysmorphic features, and congenital malformations detected an interstitial 1p22.2-p31.1 deletion in a patient with features overlapping the goldenhar syndrome. Am. J. Med. Genet. Part A 2008, 146, 2109–2115. [Google Scholar] [CrossRef]
- Spineli-Silva, S.; Sgardioli, I.C.; Dos Santos, A.P.; Bergamini, L.L.; Monlleó, I.L.; Fontes, M.I.; Félix, T.M.; Ribeiro, E.M.; Xavier, A.C.; Lustosa-Mendes, E.; et al. Genomic imbalances in craniofacial microsomia. Am. J. Med. Genet. Part C 2020, 184, 970–985. [Google Scholar] [CrossRef]
- Luquetti, D.V.; Heike, C.L.; Zarante, I.; Timms, A.E.; Gustafson, J.; Pachajoa, H.; Porras-Hurtado, G.L.; Ayala-Ramirez, P.; Duenas-Roque, M.M.; Jimenez, N.; et al. MYT1 role in the microtia-craniofacial microsomia spectrum. Mol. Genet. Genom. Med. 2020, 8, e1401. [Google Scholar] [CrossRef]
- Wang, Y.; Ping, L.; Luan, X.; Chen, Y.; Fan, X.; Li, L.; Liu, Y.; Wang, P.; Zhang, S.; Zhang, B.; et al. A Mutation in VWA1, Encoding von Willebrand Factor A Domain-Containing Protein 1, Is Associated With Hemifacial Microsomia. Front. Cell Dev. Biol. 2020, 8, 571004. [Google Scholar] [CrossRef]
- Rengasamy Venugopalan, S.; Farrow, E.; Sanchez–Lara, P.A.; Yen, S.; Lypka, M.; Jiang, S.; Allareddy, V. A novel nonsense substitution identified in the AMIGO2 gene in an Occulo-Auriculo-Vertebral spectrum patient. Orthod. Craniofacial. Res. 2019, 22, 163–167. [Google Scholar] [CrossRef]
- Zamariolli, M.; Colovati, M.; Moysés-Oliveira, M.; Nunes, N.; Caires dos Santos, L.; Alvarez Perez, A.B.; Bragagnolo, S.; Melaragno, M.I. Rare single-nucleotide variants in oculo-auriculo-vertebral spectrum (OAVS). Mol. Genet. Genom. Med. 2019, 7, e00959. [Google Scholar] [CrossRef]
- Güleray, N.; Koşukcu, C.; Oğuz, S.; Ürel Demir, G.; Taşkıran, E.Z.; Kiper, P.Ö.; Utine, G.E.; Alanay, Y.; Boduroğlu, K.; Alikaşifoğlu, M. Investigation of Genetic Causes in a Developmental Disorder: Oculoauriculovertebral Spectrum. Cleft Palate-Craniofacial. J. 2022, 59, 1114–1124. [Google Scholar] [CrossRef]
- Rengasamy Venugopalan, S.; Farrow, E.G.; Lypka, M. Whole-exome sequencing identified a variant in EFTUD2 gene in establishing a genetic diagnosis. Orthod. Craniofacial. Res. 2017, 20, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Celse, T.; Tingaud-Sequeira, A.; Dieterich, K.; Siegfried, G.; Lecaignec, C.; Bouneau, L.; Fannemel, M.; Salaun, G.; Laffargue, F.; Martinez, G.; et al. OTX2 duplications: A recurrent cause of oculo-auriculo-vertebral spectrum. J. Med. Genet. 2022. [CrossRef] [PubMed]
- Guida, V.; Sinibaldi, L.; Pagnoni, M.; Bernardini, L.; Loddo, S.; Margiotti, K.; Digilio, M.C.; Fadda, M.T.; Dallapiccola, B.; Iannetti, G.; et al. A de novo proximal 3q29 chromosome microduplication in a patient with oculo auriculo vertebral spectrum. Am. J. Med. Genet. Part A 2015, 167A, 797–801. [Google Scholar] [CrossRef]
- Puvabanditsin, S.; February, M.; Francois, L.; Garrow, E.; Bruno, C.; Mehta, R. 7q21.11 Microdeletion in a Neonate With Goldenhar Syndrome: Case Report and a Literature Review. Cleft Palate-Craniofacial. J. 2016, 53, 249–252. [Google Scholar] [CrossRef]
- Glaeser, A.B.; Santos, A.S.; Diniz, B.L.; Deconte, D.; Rosa, R.F.M.; Zen, P.R.G. Candidate genes of oculo-auriculo-vertebral spectrum in 22q region: A systematic review. Am. J. Med. Genet. Part A 2020, 182, 2624–2631. [Google Scholar] [CrossRef]
- Jordan, V.K.; Zaveri, H.P.; Scott, D.A. 1p36 deletion syndrome: An update. Appl. Clin. Genet. 2015, 8, 189–200. [Google Scholar] [PubMed]
- Kang, S.H.; Scheffer, A.; Ou, Z.; Li, J.; Scaglia, F.; Belmont, J.; Lalani, S.R.; Roeder, E.; Enciso, V.; Braddock, S.; et al. Identification of proximal 1p36 deletions using array-CGH: A possible new syndrome. Clin. Genet. 2007, 72, 329–338. [Google Scholar] [CrossRef]
- Aagaard Nolting, L.; Brasch-Andersen, C.; Cox, H.; Kanani, F.; Parker, M.; Fry, A.E.; Loddo, S.; Novelli, A.; Dentici, M.L.; Joss, S.; et al. A new 1p36.13-1p36.12 microdeletion syndrome characterized by learning disability, behavioral abnormalities, and ptosis. Clin. Genet. 2020, 97, 927–932. [Google Scholar] [CrossRef]
- Kalhori, M.R.; Soleimani, M.; Arefian, E.; Alizadeh, A.M.; Mansouri, K.; Echeverria, J. The potential role of miR-1290 in cancer progression, diagnosis, prognosis, and treatment: An oncomiR or onco-suppressor microRNA? J. Cell. Biochem. 2022, 123, 506–531. [Google Scholar] [CrossRef]
- Banerjee, A.; Jothimani, G.; Prasad, S.V.; Marotta, F.; Pathak, S. Targeting Wnt Signaling through Small molecules in Governing Stem Cell Fate and Diseases. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 233–246. [Google Scholar] [CrossRef]
- Teufel, S.; Hartmann, C. Wnt-signaling in skeletal development. Curr. Top. Dev. Biol. 2019, 133, 235–279. [Google Scholar] [PubMed]
- Li, R.; Liu, S.; Li, Y.; Tang, Q.; Xie, Y.; Zhai, R. Long noncoding RNA AFAP1-AS1 enhances cell proliferation and invasion in osteosarcoma through regulating miR-4695-5p/TCF4-β-catenin signaling. Mol. Med. Rep. 2018, 18, 1616–1622. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene Variants | Chromosome | Probands | Ref. |
|---|---|---|---|
| SF3B2 | 11q13.1 | 7 | [5] |
| MYT1 | 20q13.33 | 6 | [5,16] |
| EYA3 | 1p35.3 | 2 | [11] |
| ZYG11B | 1p32.3 | 1 | [5] |
| VWA1 | 1p36.33 | 1 | [17] |
| ZIC2 | 13q32.3 | 1 | [5] |
| AMIGO2 | 12q13.11 | 1 | [18] |
| YPEL1 | 22q11.21-22 | 2 | [19] |
| CRKL | 22q11.21 | 1 | [19] |
| OTX2 | 14q22.3 | 1 | [19] |
| PAX1 | 20p11.22 | 1 | [12] |
| EFTUD2 | 17q21.31 | 2 | [20,21] |
| Location | Probands | Ref. | Location | Probands | Ref. |
|---|---|---|---|---|---|
| 1p22-p31 del | 1 | [15] ** | 13q34 dupl | 1 | [15] ** |
| 2p12 dupl | 1 | [15] ** | 14q22.3 dupl | 10 | [22] |
| 3q29 dupl | 1 | [23] | 14q23 dup | 1 | [15] ** |
| 4p15.1 dupl | 1 | [20] | 14q31 del | 1 | [15] ** |
| 5p15 del | 1 | [15] ** | 15q24 del | 1 | [15] ** |
| 5q13.2 del | 1 | [1] | 15q26.2 del | 1 | [13] ** |
| 5q31.2 dup | 1 | [13] ** | 16p13.11 (*) | 1 | [20] |
| 7q21.11 del | 1 | [24] | 16p13.3 del | 1 | [15] ** |
| 8p22 del (*) | 1 | [20] | 17q11 dupl | 1 | [15] ** |
| 8q13.3 del | 1 | [15] ** | 22q11 del/dup | >20 | [15,25] ** |
| 10p14 dupl | 1 | [1] | 22qter del | 1 | [1] |
| 10q26.2 del | 1 | [15] ** | Xp22 del | 1 | [15] ** |
| 12p13 del | 1 | [15] ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Castro, M.; Martinez-Merino, T.; Puente, N.; Riancho, J.A. Expanding the Etiology of Oculo–Auriculo–Vertebral Spectrum: A Novel Interstitial Microdeletion at 1p36. Int. J. Mol. Sci. 2023, 24, 36. https://doi.org/10.3390/ijms24010036
García-Castro M, Martinez-Merino T, Puente N, Riancho JA. Expanding the Etiology of Oculo–Auriculo–Vertebral Spectrum: A Novel Interstitial Microdeletion at 1p36. International Journal of Molecular Sciences. 2023; 24(1):36. https://doi.org/10.3390/ijms24010036
Chicago/Turabian StyleGarcía-Castro, Mónica, Teresa Martinez-Merino, Nuria Puente, and José A. Riancho. 2023. "Expanding the Etiology of Oculo–Auriculo–Vertebral Spectrum: A Novel Interstitial Microdeletion at 1p36" International Journal of Molecular Sciences 24, no. 1: 36. https://doi.org/10.3390/ijms24010036
APA StyleGarcía-Castro, M., Martinez-Merino, T., Puente, N., & Riancho, J. A. (2023). Expanding the Etiology of Oculo–Auriculo–Vertebral Spectrum: A Novel Interstitial Microdeletion at 1p36. International Journal of Molecular Sciences, 24(1), 36. https://doi.org/10.3390/ijms24010036