

The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome

 , , and
, , and 
Abstract
1. Introduction
2. Results
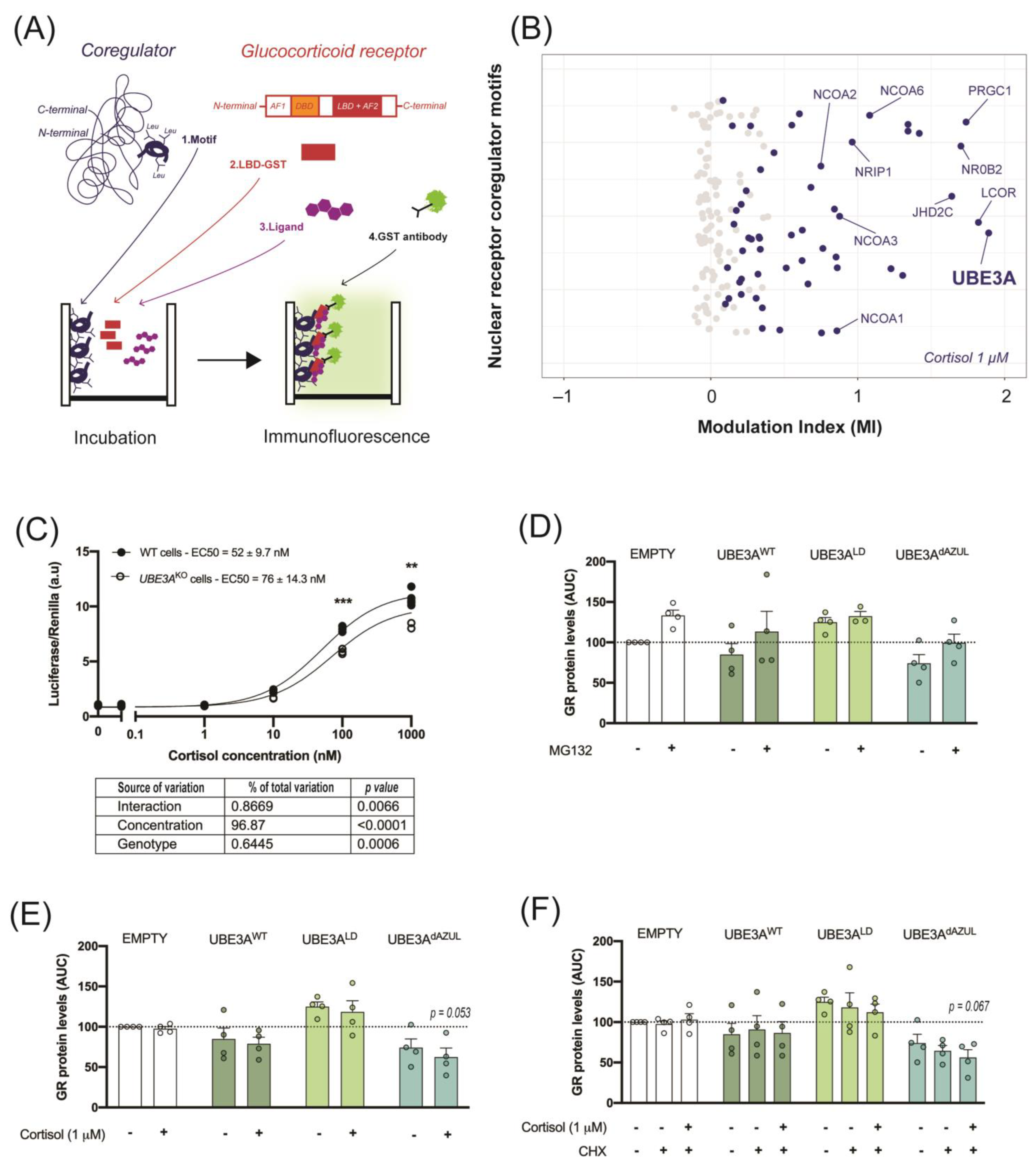
2.1. UBE3A Interacts with the GR Ligand Binding Domain
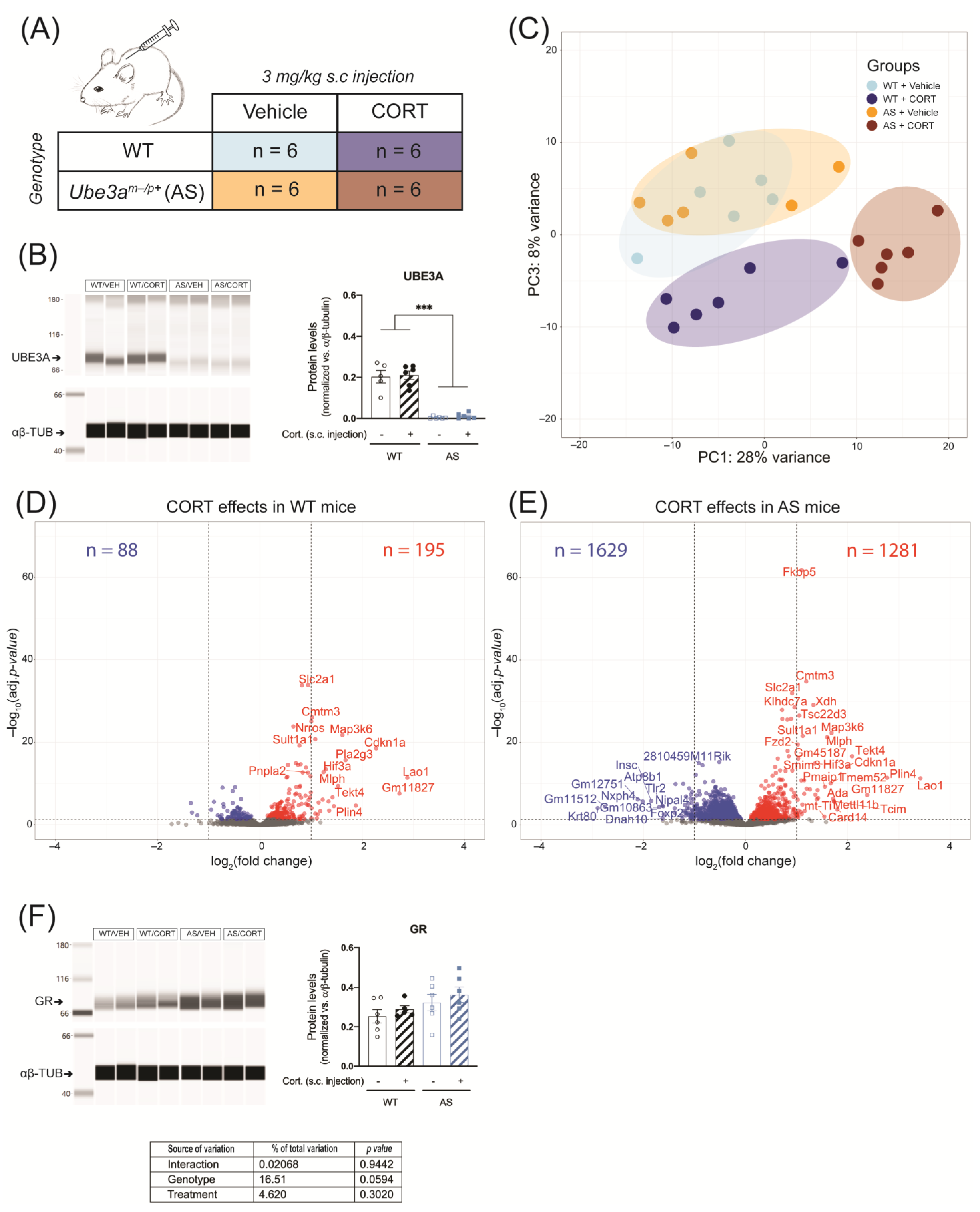
2.2. UBE3A Deficiency in AS Mice Alters Circulating Corticosterone Levels
2.3. Acute CORT Exposure Strongly Alters Hippocampal GR Signaling in UBE3A-Deficient AS Mice
2.4. Acute CORT Exposure Influences Pathways Associated with Transcription Activity and Neurotransmitter Signaling in the Hippocampus of UBE3A-Deficient AS Mice
2.5. Acute CORT Exposure Influences Genes in the UBE3A-Deficient AS Mouse Hippocampus That are Heterogeneously Expressed in Hippocampal Cell Types
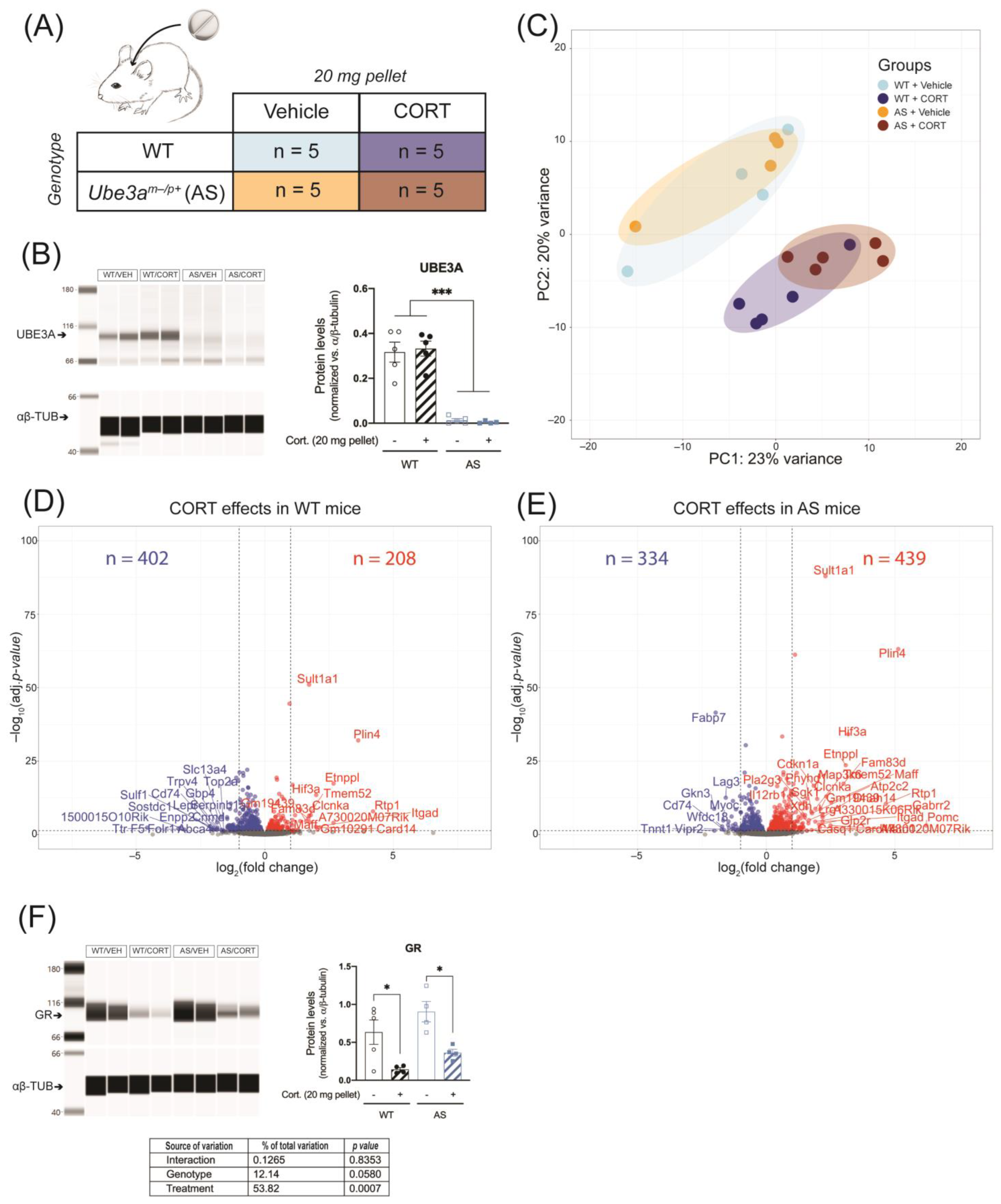
2.6. Continuous CORT Exposure Does Not Differentially Alter Hippocampal GR Signaling in UBE3A-Deficient AS Mice
2.7. Continuous CORT Exposure Slightly Alters Liver GR Signaling in UBE3A-Deficient AS Mice
3. Discussion
4. Materials and Methods
4.1. Microarray Assay for Realtime Coregulator-Nuclear Receptor Interaction (MARCoNI)
4.2. Cell Culture
4.2.1. GR Activity
4.2.2. GR Stability
4.3. Animals
4.3.1. Acute Corticosterone Exposure
4.3.2. Continuous Corticosterone Exposure
4.4. Corticosterone Biochemical Analysis
4.5. Protein Levels Measurements in Mouse Brain and Liver Tissue
4.6. RNA Sequencing (RNA-seq) Analysis of Mouse Hippocampus and Liver Tissue
4.7. Pathway Enrichment Analysis
4.8. Single-Cell RNA Sequencing in the Adult Mouse Hippocampus
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF1, AF2 | Activation function domain 1 and 2 |
| αβ-TUB—αβ-tubulin | Alpha-beta Tubulin |
| ANOVA | Analysis of variance |
| AR | Androgen receptor |
| AS | Angelman syndrome |
| CA1-ProS, CA2, CA3 | Hippocampal Cornus ammonis regions 1, 2, 3 |
| CHX | Cycloheximide |
| CORT | Corticosterone |
| DBD | DNA binding domain |
| DG | Dentate Gyrus |
| DMSO | Dimethyl-sulfoxide |
| ERα, ERβ | Estrogen receptor α and β |
| FBS | Fetal Bovine Serum |
| FCS | Fetal Calf Serum |
| FDR | False discovery rate |
| GO | Gene ontology |
| GR | Glucocorticoid receptor |
| GRE | Glucocorticoid response elements |
| GST | Glutathione S-transferase |
| HPA axis | Hypothalamic pituitary adrenal axis |
| KO | knock-out |
| LBD | Ligand binding domain |
| LxxLL | Short leucine-rich binding motifs |
| MARCoNI | Microarray Assay for Realtime Coregulator-Nuclear receptor Interaction |
| MI | Modulation Index |
| NR-box | Nuclear receptor box |
| p | p-value |
| PBS | Phosphate-buffered saline |
| PCA | Principal Component Analysis |
| PC | Principal component |
| PR | Progesterone receptor |
| PVN | Paraventricular nucleus |
| RNA-seq | RNA sequencing |
| scRNA-seq | Single-cell RNA sequencing |
| s.c. | subcutaneous |
| SD | standard deviation |
| SEM | Standard error of the mean |
| TAT | Tyrosine aminotransferase |
| UBE3A | Ubiquitin protein ligase E3A |
| UBE3AWT | Wildtype UBE3A |
| UBE3ALD | Ligase-dead UBE3A mutant |
| UBE3AdAZUL | dAZUL UBE3A mutant |
| WT | Wildtype |
References
- Williams, C.A. The behavioral phenotype of the Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 432–437. [Google Scholar] [CrossRef]
- Buiting, K. Prader–Willi syndrome and Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef]
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef]
- Matsuura, T.; Sutcliffe, J.S.; Fang, P.; Galjaard, R.J.; Jiang, Y.H.; Benton, C.S.; Rommens, J.M.; Beaudet, A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997, 15, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, U.; Sutcliffe, J.S.; Cattanach, B.M.; Beechey, C.V.; Armstrong, D.; Eichele, G.; Beaudet, A.L. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 1997, 17, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Jiang, Y.H.; Galjaard, R.J.; Matsuura, T.; Fang, P.; Kubota, T.; Christian, S.L.; Bressler, J.; Cattanach, B.; Ledbetter, D.H.; et al. The E6–AP Ubiquitin–Protein Ligase (UBE3A) Gene Is Localized within a Narrowed Angelman Syndrome Critical Region. Genome Res. 1997, 7, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Hoffman, A.R. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 1997, 17, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Joh, K.; Ohta, T.; Masuzaki, H.; Ishimaru, T.; Mukai, T.; Niikawa, N.; Ogawa, M.; Wagstaff, J.; Kishino, T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet. 2003, 12, 837–847. [Google Scholar] [CrossRef]
- Lossie, A.C.; Whitney, M.M.; Amidon, D.; Dong, H.J.; Chen, P.; Theriaque, D.; Hutson, A.; Nicholls, R.D.; Zori, R.T.; Williams, C.A.; et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 2001, 38, 834–845. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567. [Google Scholar] [CrossRef]
- Scheffner, M.; Number, U.; Huibregtse, J.M. Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef]
- Cooper, E.M.; Hudson, A.W.; Amos, J.; Wagstaff, J.; Howley, P.M. Biochemical Analysis of Angelman Syndrome-associated Mutations in the E3 Ubiquitin Ligase E6-associated Protein. J. Biol. Chem. 2004, 279, 41208–41217. [Google Scholar] [CrossRef]
- Bossuyt, S.N.; Punt, A.M.; De Graaf, I.J.; Van Den Burg, J.; Williams, M.G.; Heussler, H.; Elgersma, Y.; Distel, B. Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum. Mol. Genet. 2021, 30, 430–442. [Google Scholar] [CrossRef]
- Avagliano Trezza, R.; Sonzogni, M.; Bossuyt, S.N.; Zampeta, F.I.; Punt, A.M.; van den Berg, M.; Rotaru, D.C.; Koene, L.; Munshi, S.T.; Stedehouder, J.; et al. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 2019, 22, 1235–1247. [Google Scholar] [CrossRef]
- Jason, J.Y.; Berrios, J.; Newbern, J.M.; Snider, W.D.; Philpot, B.D.; Hahn, K.M.; Zylka, M.J. An Autism-Linked Mutation Disables Phosphorylation Control of UBE3A. Cell 2015, 162, 795–807. [Google Scholar] [CrossRef]
- Burris, T.P.; Nawaz, Z.; Tsai, M.J.; O’Malley, B.W. A nuclear hormone receptor-associated protein that inhibits transactivation by the thyroid hormone and retinoic acid receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 9525–9529. [Google Scholar] [CrossRef]
- Nawaz, Z.; Lonard, D.M.; Smith, C.L.; Lev-Lehman, E.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. The Angelman Syndrome-Associated Protein, E6-AP, Is a Coactivator for the Nuclear Hormone Receptor Superfamily. Mol. Cell Biol. 1999, 19, 1182–1189. [Google Scholar] [CrossRef]
- Catoe, H.W.; Nawaz, Z. E6-AP facilitates efficient transcription at estrogen responsive promoters through recruitment of chromatin modifiers. Steroids 2011, 76, 897–902. [Google Scholar] [CrossRef] [PubMed]
- El Hokayem, J.; Nawaz, Z. E6AP in the Brain: One Protein, Dual Function, Multiple Diseases. Mol. Neurobiol. 2014, 49, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Joëls, M. Corticosteroids and the brain. J. Endocrinol. 2018, 238, R121–R130. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Joëls, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef]
- Viho, E.M.; Buurstede, J.C.; Mahfouz, A.; Koorneef, L.L.; Van Weert, L.T.; Houtman, R.; Hunt, H.J.; Kroon, J.; Meijer, O.C. Corticosteroid Action in the Brain: The Potential of Selective Receptor Modulation. Neuroendocrinology 2019, 109, 266–276. [Google Scholar] [CrossRef]
- Goldfarb, E.V.; Rosenberg, M.D.; Seo, D.; Constable, R.T.; Sinha, R. Hippocampal seed connectome-based modeling predicts the feeling of stress. Nat. Commun. 2020, 11, 2650. [Google Scholar] [CrossRef]
- Spiga, F.; Walker, J.J.; Terry, J.R.; Lightman, S.L. HPA Axis-Rhythms. Compr. Physiol. 2014, 4, 1273–1298. [Google Scholar]
- de Kloet, E.R.; Sarabdjitsingh, R.A. Everything Has Rhythm: Focus on Glucocorticoid Pulsatility. Endocrinology 2008, 149, 3241–3243. [Google Scholar] [CrossRef]
- Fitzsimons, C.P.; Herbert, J.; Schouten, M.; Meijer, O.C.; Lucassen, P.J.; Lightman, S. Circadian and ultradian glucocorticoid rhythmicity: Implications for the effects of glucocorticoids on neural stem cells and adult hippocampal neurogenesis. Front. Neuroendocrinol. 2016, 41, 44–58. [Google Scholar] [CrossRef]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- Mahfouz, A.; Lelieveldt, B.P.; Grefhorst, A.; Van Weert, L.T.; Mol, I.M.; Sips, H.C.; Van Den Heuvel, J.K.; Datson, N.A.; Visser, J.A.; Reinders, M.J.; et al. Genome-wide coexpression of steroid receptors in the mouse brain: Identifying signaling pathways and functionally coordinated regions. Proc. Natl. Acad. Sci. USA 2016, 113, 2738–2743. [Google Scholar] [CrossRef]
- Viho, E.M.; Buurstede, J.C.; Berkhout, J.B.; Mahfouz, A.; Meijer, O.C. Cell type specificity of glucocorticoid signaling in the adult mouse hippocampus. J. Neuroendocrinol. 2022, 34, e13072. [Google Scholar] [CrossRef]
- Godavarthi, S.K.; Dey, P.; Maheshwari, M.; Ranjan Jana, N. Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum. Mol. Genet. 2012, 21, 1824–1834. [Google Scholar] [CrossRef]
- Kim, J.; Lee, B.; Kim, D.H.; Yeon, J.G.; Lee, J.; Park, Y.; Lee, Y.; Lee, S.K.; Lee, S.; Lee, J.W. UBE3A Suppresses Overnutrition-Induced Expression of the Steatosis Target Genes of MLL4 by Degrading MLL4. Hepatology 2019, 69, 1122–1134. [Google Scholar] [CrossRef]
- Shi, S.Q.; Bichell, T.J.; Ihrie, R.A.; Johnson, C.H. Ube3a Imprinting Impairs Circadian Robustness in Angelman Syndrome Models. Curr. Biol. 2015, 25, 537–545. [Google Scholar] [CrossRef]
- Koorneef, L.L.; van den Heuvel, J.K.; Kroon, J.; Boon, M.R.; ’t Hoen, P.A.; Hettne, K.M.; van de Velde, N.M.; Kolenbrander, K.B.; Streefland, T.C.; Mol, I.M.; et al. Selective Glucocorticoid Receptor Modulation Prevents and Reverses Nonalcoholic Fatty Liver Disease in Male Mice. Endocrinology 2018, 159, 3925–3936. [Google Scholar] [CrossRef]
- Kroon, J.; Schilperoort, M.; het Panhuis, W.I.; van den Berg, R.; van Doeselaar, L.; Verzijl, C.R.; van Trigt, N.; Mol, I.M.; Sips, H.H.; van den Heuvel, J.K.; et al. A physiological glucocorticoid rhythm is an important regulator of brown adipose tissue function. Mol. Metab. 2021, 47, 101179. [Google Scholar] [CrossRef]
- Rahimi, L.; Rajpal, A.; Ismail-Beigi, F. Glucocorticoid-Induced Fatty Liver Disease. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1133–1145. [Google Scholar] [CrossRef]
- Zampeta, F.I.; Sonzogni, M.; Niggl, E.; Lendemeijer, B.; Smeenk, H.; de Vrij, F.M.; Kushner, S.A.; Distel, B.; Elgersma, Y. Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum. Mol. Genet. 2020, 29, 3032–3043. [Google Scholar] [CrossRef]
- Broekema, M.F.; Hollman, D.A.; Koppen, A.; van den Ham, H.J.; Melchers, D.; Pijnenburg, D.; Ruijtenbeek, R.; van Mil, S.W.; Houtman, R.; Kalkhoven, E. Profiling of 3696 Nuclear Receptor–Coregulator Interactions: A Resource for Biological and Clinical Discovery. Endocrinology 2018, 159, 2397–2407. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Nawaz, Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl. Recept. Signal. 2008, 6, nrs06006. [Google Scholar] [CrossRef] [PubMed]
- Spencer, R.L.; Deak, T. A users guide to HPA axis research. Physiol. Behav. 2017, 178, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Wolfer, D.P.; Selbach, O.; Alter, H.; Erdmann, G.; Reichardt, H.M.; Chepkova, A.N.; Welzl, H.; Haas, H.L.; Lipp, H.P.; et al. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc. Natl. Acad. Sci. USA 2006, 103, 195–200. [Google Scholar] [CrossRef] [PubMed]
- ter Heegde, F.; De Rijk, R.H.; Vinkers, C.H. The brain mineralocorticoid receptor and stress resilience. Psychoneuroendocrinology 2015, 52, 92–110. [Google Scholar] [CrossRef]
- Lightman, S.L.; Birnie, M.T.; Conway-Campbell, B.L. Dynamics of ACTH and Cortisol Secretion and Implications for Disease. Endocr. Rev. 2020, 41, bnaa002. [Google Scholar] [CrossRef]
- Ratka, A.; Sutanto, W.; Bloemers, M.; de Kloet, R. On the Role of Brain Mineralocorticoid (Type I) and Glucocorticoid (Type II) Receptors in Neuroendocrine Regulation. Neuroendocrinology 1989, 50, 117–123. [Google Scholar] [CrossRef]
- Aguilera, C.; Gabau, E.; Ramirez-Mallafré, A.; Brun-Gasca, C.; Dominguez-Carral, J.; Delgadillo, V.; Laurie, S.; Derdak, S.; Padilla, N.; de la Cruz, X.; et al. New genes involved in Angelman syndrome-like: Expanding the genetic spectrum. PLoS ONE 2021, 16, e0258766. [Google Scholar] [CrossRef]
- Farook, M.F.; DeCuypere, M.; Hyland, K.; Takumi, T.; LeDoux, M.S.; Reiter, L.T. Altered Serotonin, Dopamine and Norepinepherine Levels in 15q Duplication and Angelman Syndrome Mouse Models. PLoS ONE 2012, 7, e43030. [Google Scholar] [CrossRef]
- Kaphzan, H.; Buffington, S.A.; Jung, J.I.; Rasband, M.N.; Klann, E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J. Neurosci. Off J. Soc. Neurosci. 2011, 31, 17637–17648. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman Ubiquitin Ligase in Mice Causes Increased Cytoplasmic p53 and Deficits of Contextual Learning and Long-Term Potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef]
- Rotaru, D.C.; van Woerden, G.M.; Wallaard, I.; Elgersma, Y. Adult Ube3a Gene Reinstatement Restores the Electrophysiological Deficits of Prefrontal Cortex Layer 5 Neurons in a Mouse Model of Angelman Syndrome. J. Neurosci. 2018, 38, 8011–8030. [Google Scholar] [CrossRef]
- Silva-Santos, S.; Van Woerden, G.M.; Bruinsma, C.F.; Mientjes, E.; Jolfaei, M.A.; Distel, B.; Kushner, S.A.; Elgersma, Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest. 2015, 125, 2069–2076. [Google Scholar] [CrossRef]
- Judson, M.C.; Wallace, M.L.; Sidorov, M.S.; Burette, A.C.; Gu, B.; van Woerden, G.M.; King, I.F.; Han, J.E.; Zylka, M.J.; Elgersma, Y.; et al. GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron 2016, 90, 56–69. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, G.; Liu, Y.; Standley, S.; Ji, A.; Tunuguntla, R.; Wang, Y.; Claus, C.; Luo, Y.; Baudry, M.; et al. UBE3A Regulates Synaptic Plasticity and Learning and Memory by Controlling SK2 Channel Endocytosis. Cell Rep. 2015, 12, 449–461. [Google Scholar] [CrossRef]
- Egawa, K.; Kitagawa, K.; Inoue, K.; Takayama, M.; Takayama, C.; Saitoh, S.; Kishino, T.; Kitagawa, M.; Fukuda, A. Decreased Tonic Inhibition in Cerebellar Granule Cells Causes Motor Dysfunction in a Mouse Model of Angelman Syndrome. Sci. Transl. Med. 2012, 4, 163ra157. [Google Scholar] [CrossRef]
- Margolis, S.S.; Salogiannis, J.; Lipton, D.M.; Mandel-Brehm, C.; Wills, Z.P.; Mardinly, A.R.; Hu, L.; Greer, P.L.; Bikoff, J.B.; Ho, H.Y.H.; et al. EphB-Mediated Degradation of the RhoA GEF Ephexin5 Relieves a Developmental Brake on Excitatory Synapse Formation. Cell 2010, 143, 442–455. [Google Scholar] [CrossRef]
- Kaphzan, H.; Buffington, S.A.; Ramaraj, A.B.; Lingrel, J.B.; Rasband, M.N.; Santini, E.; Klann, E. Genetic Reduction of the α1 Subunit of Na/K-ATPase Corrects Multiple Hippocampal Phenotypes in Angelman Syndrome. Cell Rep. 2013, 4, 405–412. [Google Scholar] [CrossRef]
- Judson, M.C.; Sosa-Pagan, J.O.; Del Cid, W.A.; Han, J.E.; Philpot, B.D. Allelic specificity of Ube3a Expression in The Mouse Brain During Postnatal Development. J. Comp. Neurol. 2014, 522, 1874–1896. [Google Scholar] [CrossRef]
- Flati, T.; Gioiosa, S.; Chillemi, G.; Mele, A.; Oliverio, A.; Mannironi, C.; Rinaldi, A.; Castrignanò, T. A gene expression atlas for different kinds of stress in the mouse brain. Sci. Data 2020, 7, 437. [Google Scholar] [CrossRef]
- Filipović, D.; Zlatković, J.; Gass, P.; Inta, D. The differential effects of acute vs. chronic stress and their combination on hippocampal parvalbumin and inducible heat shock protein 70 expression. Neuroscience 2013, 236, 47–54. [Google Scholar] [CrossRef]
- Wheeler, A.C.; Sacco, P.; Cabo, R. Unmet clinical needs and burden in Angelman syndrome: A review of the literature. Orphanet. J. Rare Dis. 2017, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.C.; Okoniewski, K.C.; Wylie, A.; DeRamus, M.; Hiruma, L.S.; Toth, D.; Christian, R.B. Anxiety-associated and separation distress-associated behaviours in Angelman syndrome. J. Intellect. Disabil. Res. 2019, 63, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Gentile, J.K.; Tan, W.H.; Horowitz, L.T.; Bacino, C.A.; Skinner, S.A.; Barbieri-Welge, R.; Bauer-Carlin, A.; Beaudet, A.L.; Bichell, T.J.; Lee, H.S.; et al. A Neurodevelopmental Survey of Angelman Syndrome with Genotype-Phenotype Correlations. J. Dev. Behav. Pediatr. 2010, 31, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Wink, L.K.; Fitzpatrick, S.; Shaffer, R.; Melnyk, S.; Begtrup, A.H.; Fox, E.; Schaefer, T.L.; Mathieu-Frasier, L.; Ray, B.; Lahiri, D.; et al. The neurobehavioral and molecular phenotype of Angelman Syndrome. Am. J. Med. Genet. A 2015, 167, 2623–2628. [Google Scholar] [CrossRef] [PubMed]
- Keary, C.J.; Mullett, J.E.; Nowinski, L.; Wagner, K.; Walsh, B.; Saro, H.K.; Erhabor, G.; Thibert, R.L.; McDougle, C.J.; Ravichandran, C.T. Parent Description of Anxiety in Angelman Syndrome. J. Autism Dev. Disord. 2022, 52, 3612–3625. [Google Scholar] [CrossRef]
- Desmet, S.J.; Dejager, L.; Clarisse, D.; Thommis, J.; Melchers, D.; Bastiaensen, N.; Ruijtenbeek, R.; Beck, I.M.; Libert, C.; Houtman, R.; et al. Cofactor Profiling of the Glucocorticoid Receptor from a Cellular Environment. In Steroid Receptors; Castoria, G., Auricchio, F., Eds.; Humana: New York, NY, USA, 2014; pp. 83–94. [Google Scholar]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef]
- Liu, W.; Wang, J.; Yu, G.; Pearce, D. Steroid receptor transcriptional synergy is potentiated by disruption of the DNA-binding domain dimer interface. Mol. Endocrinol. 1996, 10, 1399–1406. [Google Scholar] [CrossRef]
- Buurstede, J.C.; Umeoka, E.H.; da Silva, M.S.; Krugers, H.J.; Joels, M.; Meijer, O.C. Application of a pharmacological transcriptome filter identifies a shortlist of mouse glucocorticoid receptor target genes associated with memory consolidation. Neuropharmacology 2022, 216, 109186. [Google Scholar] [CrossRef]
- Buurstede, J.C.; van Weert, L.T.; Colucci, P.; Gentenaar, M.; Viho, E.M.; Koorneef, L.L.; Schoonderwoerd, R.A.; Lanooij, S.D.; Moustakas, I.; Balog, J.; et al. Hippocampal glucocorticoid target genes associated with enhancement of memory consolidation. Eur. J. Neurosci. 2022, 55, 2666–2683. [Google Scholar] [CrossRef]
- Atucha, E.; Zalachoras, I.; van den Heuvel, J.K.; van Weert, L.T.; Melchers, D.; Mol, I.M.; Belanoff, J.K.; Houtman, R.; Hunt, H.; Roozendaal, B.; et al. A Mixed Glucocorticoid/Mineralocorticoid Selective Modulator with Dominant Antagonism in the Male Rat Brain. Endocrinology 2015, 156, 4105–4114. [Google Scholar] [CrossRef]
- Spaanderman, D.C.; Nixon, M.; Buurstede, J.C.; Sips, H.H.; Schilperoort, M.; Kuipers, E.N.; Backer, E.A.; Kooijman, S.; Rensen, P.C.; Homer, N.Z.; et al. Androgens modulate glucocorticoid receptor activity in adipose tissue and liver. J. Endocrinol. 2019, 240, 51–63. [Google Scholar] [CrossRef]
- Viho, E.M.; Kroon, J.; Feelders, R.A.; Houtman, R.; van den Dungen, E.; Arias, A.M.P.; Hunt, H.; Hofland, L.J.; Meijer, O.C. Peripheral glucocorticoid receptor antagonism by relacorilant with modest HPA axis disinhibition. J. Endocrinol. 2022, 1. [Google Scholar] [CrossRef]
- Koorneef, L.L.; Kroon, J.; Viho, E.M.; Wahl, L.F.; Heckmans, K.M.; van Dorst, M.M.; Hoekstra, M.; Houtman, R.; Hunt, H.; Meijer, O.C. The selective glucocorticoid receptor antagonist CORT125281 has tissue-specific activity. J. Endocrinol. 2020, 246, 79–92. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Target | Antibody Species | Supplier | Product Number | Dilution |
|---|---|---|---|---|
| Primary Antibodies | ||||
| Anti-GR | Rabbit | Cell Signaling Technology, Danvers, MA, USA | 12041S | 1:1000 |
| Anti-β-Actin | Mouse | Chemicon Sigma-Aldrich, St. Louis, MO, USA | MAB1501R | 1:20,000 |
| Anti-GAPDH | Rabbit | Cell Signaling Technology, Danvers, MA, USA | 2118S | 1:1,000 |
| Secondary Antibodies | ||||
| Anti-mouse | Goat | LI-COR, Lincoln, NE, USA | 926-32210 | 1:15,000 |
| Anti-rabbit | Goat | LI-COR, Lincoln, NE, USA | 926-68071 | 1:15,000 |
| Antibody Target | Antibody Species | Supplier | Product Number | Dilution |
|---|---|---|---|---|
| Anti-GR | Rabbit | Cell Signaling Technology, Danvers, MA, USA | 12041S | 1:20 |
| Anti-UBE3A (for brain tissue) | Mouse | Sigma-Aldrich, St. Louis, MO, USA | E8655 | 1:20 |
| Anti-UBE3A (for liver tissue) | Mouse | Santa Cruz Biotechnology, Dallas, TX, USA | sc-166689 | 1:20 |
| Anti-αβ-tubulin | Rabbit | Cell Signaling Technology, Danvers, MA, USA | 2148S | 1:20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viho, E.M.G.; Punt, A.M.; Distel, B.; Houtman, R.; Kroon, J.; Elgersma, Y.; Meijer, O.C. The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. Int. J. Mol. Sci. 2023, 24, 303. https://doi.org/10.3390/ijms24010303
Viho EMG, Punt AM, Distel B, Houtman R, Kroon J, Elgersma Y, Meijer OC. The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. International Journal of Molecular Sciences. 2023; 24(1):303. https://doi.org/10.3390/ijms24010303
Chicago/Turabian StyleViho, Eva M. G., A. Mattijs Punt, Ben Distel, René Houtman, Jan Kroon, Ype Elgersma, and Onno C. Meijer. 2023. "The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome" International Journal of Molecular Sciences 24, no. 1: 303. https://doi.org/10.3390/ijms24010303
APA StyleViho, E. M. G., Punt, A. M., Distel, B., Houtman, R., Kroon, J., Elgersma, Y., & Meijer, O. C. (2023). The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. International Journal of Molecular Sciences, 24(1), 303. https://doi.org/10.3390/ijms24010303