
De Novo Asp219Val Mutation in Cardiac Tropomyosin Associated with Hypertrophic Cardiomyopathy
 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Results
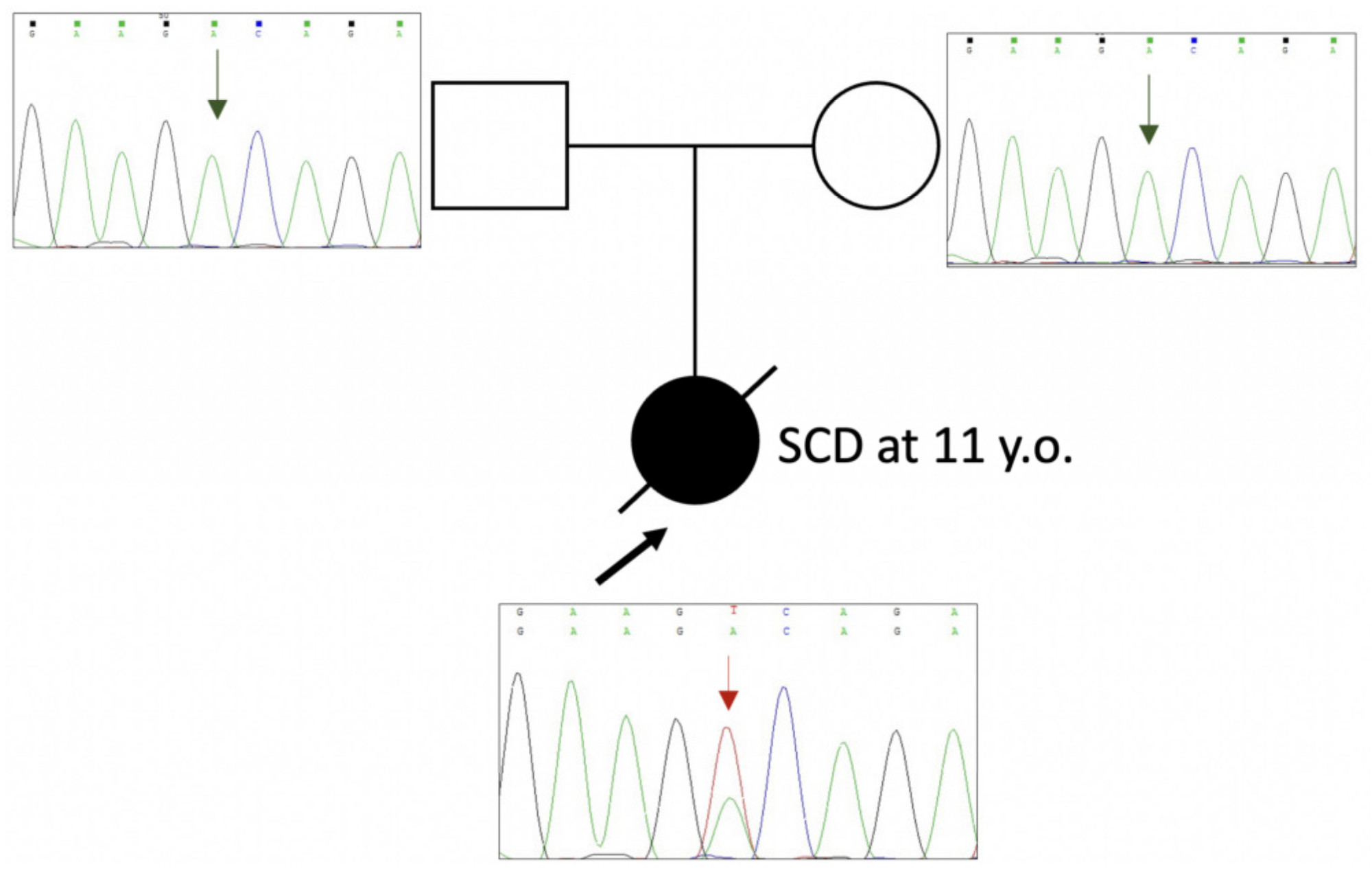
2.1. Genetic Evaluation and Mutation Detection
2.2. Effect of the D219V Mutation on Tpm Thermal Stability
2.3. Effect of the D219V Mutation on Tpm Solution Viscosity
2.4. Effect of the D219V Mutation on Tpm Binding to F-Actin
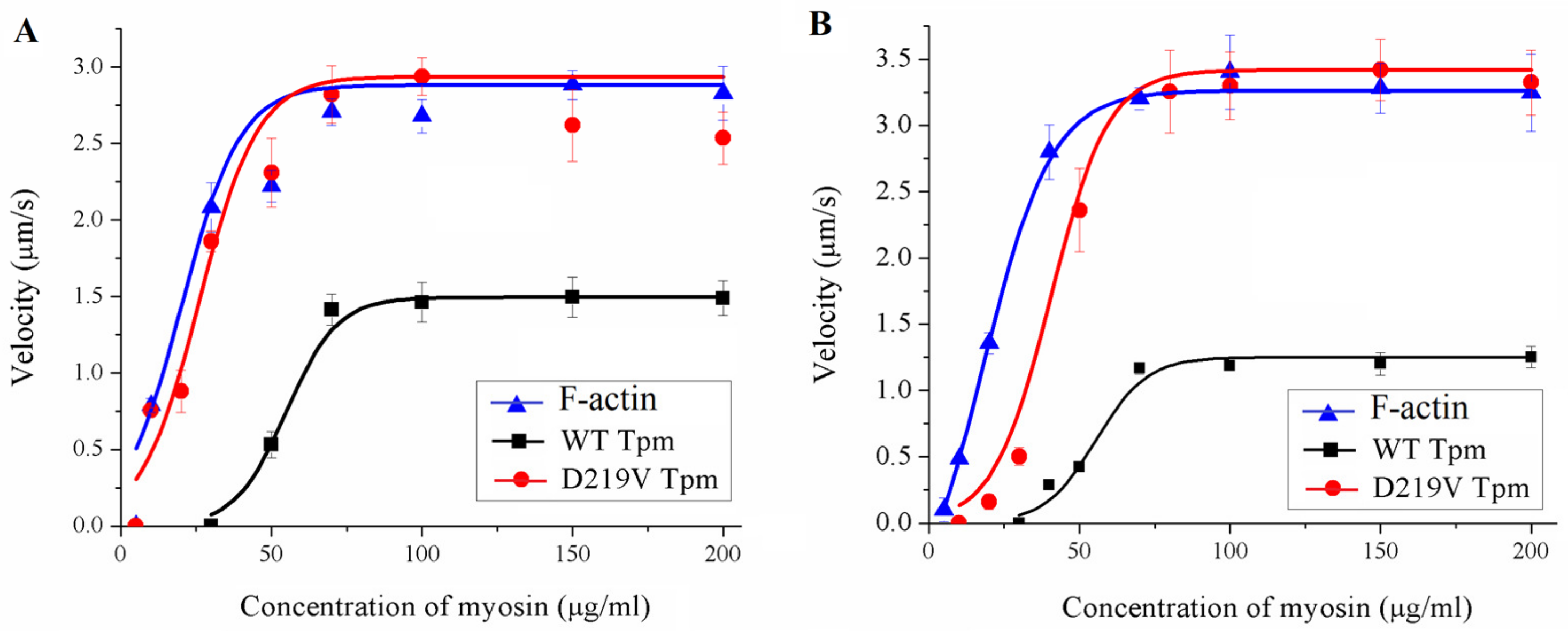
2.5. Effect of the D219V Tpm Mutation on the Actin-Myosin Interaction and Its Ca2+ Regulation In Vitro
2.6. Molecular Dynamics Simulation
3. Discussion
3.1. The D219V Tpm Mutation Increases Ca2+ Sensitivity and Impairs Relaxation of the Regulated Thin Filaments
3.2. Possible Molecular Mechanisms of the Effects of the D219V Tpm Mutation
3.3. Changes in Structure-Function Relationship Caused by the D219V Tpm Mutation
3.4. Relation to Previous Works
3.5. Limitation of the Work
4. Materials and Methods
4.1. Genetic Investigation
4.2. Protein Preparations
4.3. Differential Scanning Calorimetry (DSC)
4.4. Viscosimetry
4.5. Cosedimentation of Tpm Species with F-Actin
4.6. Temperature Dependences of Light Scattering
4.7. In Vitro Motility Assay
4.8. Molecular Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Toste, A. Advances in hypertrophic cardiomyopathy: What the cardiologist needs to know. Port. J. Cardiol. 2022, 41, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Ellsworth, E.G.; Ommen, S.R.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 2003, 108, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Ho, C.Y.; Ashley, E.; Day, S.; Ferrantini, C.; Girolami, F.; Tomberli, B.; Bardi, S.; Torricelli, F.; Cecchi, F.; et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J. Am. Coll. Cardiol. 2014, 64, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Silva Enciso, J.; Tardiff, J.C.; Adler, E.D. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, J.C. Thin filament mutations: Developing an integrative approach to a complex disorder. Circ. Res. 2011, 108, 765–782. [Google Scholar] [CrossRef]
- Marston, S.B. How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res. 2011, 4, 245–255. [Google Scholar] [CrossRef]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Lehrer, S.S.; Geeves, M.A. The myosin-activated thin filament regulatory state, M−-open: A link to hypertrophic cardiomyopathy (HCM). J. Muscle Res. Cell Motil. 2014, 35, 153–160. [Google Scholar] [CrossRef]
- Duncker, D.J.; Bakkers, J.; Brundel, B.J.; Robbins, J.; Tardiff, J.C.; Carrier, L. Animal and in silico models for the study of sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 439–448. [Google Scholar] [CrossRef]
- Marston, S.B. Why is there a Limit to the Changes in Myofilament Ca2+-Sensitivity Associated with Myopathy Causing Mutations? Front. Physiol. 2016, 7, 415. [Google Scholar] [CrossRef]
- Sequeira, V.; Bertero, E.; Maack, C. Energetic drain driving hypertrophic cardiomyopathy. FEBS Lett. 2019, 593, 1616–1626. [Google Scholar] [CrossRef]
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Levitsky, D.I. Molecular Mechanisms of Pathologies of Skeletal and Cardiac Muscles Caused by Point Mutations in the Tropomyosin Genes. Biochemistry 2020, 85 (Suppl. 1), S20–S33. [Google Scholar] [CrossRef]
- Nevzorov, I.A.; Levitsky, D.I. Tropomyosin: Double helix from the protein world. Biochemistry 2011, 76, 1507–1527. [Google Scholar] [CrossRef]
- Lehman, W. Thin Filament Structure and the Steric Blocking Model. Compar. Physiol. 2016, 6, 1043–1069. [Google Scholar] [CrossRef]
- Hitchcock-DeGregori, S.E.; Barua, B. Tropomyosin Structure, Function, and Interactions: A Dynamic Regulator. Subcell. Biochem. 2017, 82, 253–284. [Google Scholar] [CrossRef]
- Kopylova, G.V.; Matyushenko, A.M.; Koubassova, N.A.; Shchepkin, D.V.; Bershitsky, S.Y.; Levitsky, D.I.; Tsaturyan, A.K. Functional outcomes of structural peculiarities of striated muscle tropomyosin. J. Muscle Res. Cell Motil. 2020, 41, 55–70. [Google Scholar] [CrossRef]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Aguilera, B.; Banner, J.; Cohle, S.; d’Amati, G.; de Gouveia, R.H.; di Gioia, C.; Fabre, A.; Gallagher, P.J.; Leone, O.; et al. Association for European Cardiovascular Pathology. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017, 471, 691–705. [Google Scholar] [CrossRef] [PubMed]
- gnomAD Database. Available online: https://gnomad.broadinstitute.org/gene/ENSG00000140416?dataset=gnomad_r2_1 (accessed on 2 November 2022).
- Varsome Resource. Available online: https://varsome.com/variant/hg38/TPM1(NM_001365777.1)%3AD219V?annotation-mode=germline (accessed on 2 November 2022).
- Freire, E.; Biltonen, R.L. Statistical mechanical deconvolution of thermal transitions in macromolecules. I. Theory and application to homogeneous systems. Biopolymers 1978, 17, 463–479. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Artemova, N.V.; Sluchanko, N.N.; Levitsky, D.I. Effects of two stabilizing substitutions, D137L and G126R, in the middle part of α-tropomyosin on the domain structure of its molecule. Biophys. Chem. 2015, 196, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Matyushenko, A.M.; Artemova, N.V.; Shchepkin, D.V.; Kopylova, G.V.; Bershitsky, S.Y.; Tsaturyan, A.K.; Sluchanko, N.N.; Levitsky, D.I. Structural and functional effects of two stabilizing substitutions, D137L and G126R, in the middle part of α-tropomyosin molecule. FEBS J. 2014, 281, 2004–2016. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Popruga, K.E.; Artemova, N.V.; Pivovarova, A.V.; Bershitsky, S.Y.; Levitsky, D.I. Structural and functional effects of cardiomyopathy-causing mutations in troponin T-binding region of cardiac tropomyosin. Biochemistry 2017, 56, 250–259. [Google Scholar] [CrossRef]
- Shchepkin, D.V.; Nabiev, S.R.; Kopylova, G.V.; Matyushenko, A.M.; Levitsky, D.I.; Bershitsky, S.Y.; Tsaturyan, A.K. Cooperativity of myosin interaction with thin filaments is enhanced by stabilizing substitutions in tropomyosin. J. Muscle Res. Cell Motil. 2017, 38, 183–191. [Google Scholar] [CrossRef]
- VanBuren, P.; Palmiter, K.A.; Warshaw, D.M. Tropomyosin directly modulates actomyosin mechanical performance at the level of a single actin filament. Proc. Natl. Acad. Sci. USA 1999, 96, 12488–12493. [Google Scholar] [CrossRef]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef]
- Dmour, B.-A.; Miftode, R.-S.; Iliescu Halitchi, D.; Anton-Paduraru, D.T.; Iliescu Halitchi, C.-O.; Miftode, I.-L.; Mitu, O.; Costache, A.-D.; Stafie, C.-S.; Costache, I.I. Latest insights into mechanisms behind atrial cardiomyopathy: It is not always about ventricular function. Diagnostics 2021, 11, 449. [Google Scholar] [CrossRef]
- Packer, M. Characterization, pathogenesis, and clinical implications of inflammation-related atrial myopathy as an important cause of atrial fibrillation. J. Am. Heart Assoc. 2020, 9, e015343. [Google Scholar] [CrossRef]
- Carlisle, M.A.; Fudim, M.; DeVore, A.D.; Piccini, J.P. Heart failure and atrial fibrillation, like fire and fury. JACC Heart Fail. 2019, 7, 447–456. [Google Scholar] [CrossRef]
- Holeman, T.A.; Lynn, M.L.; Tardiff, J.C. Unique Structural and Functional Effects of Alpha-Tropomyosin Mutations in HCM and DCM. Biophys. J. 2018, 114, 498a. [Google Scholar] [CrossRef]
- Lehman, W.; Li, X.; Kiani, F.A.; Moore, J.R.; Campbell, S.G.; Fischer, S.; Rynkiewicz, M.J. Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophys. J. 2018, 115, 1082–1092. [Google Scholar] [CrossRef]
- Barua, B.; Pamula, M.C.; Hitchcock-DeGregori, S.E. Evolutionarily conserved surface residues constitute actin binding sites of tropomyosin. Proc. Natl. Acad. Sci. USA 2011, 108, 10150–10155. [Google Scholar] [CrossRef]
- Risi, C.; Schäfer, L.U.; Belknap, B.; Pepper, I.; White, H.D.; Schröder, G.F.; Galkin, V.E. High-Resolution Cryo-EM Structure of the Cardiac Actomyosin Complex. Structure 2021, 29, 50–60.e4. [Google Scholar] [CrossRef]
- Baldo, A.P.; Tardiff, J.C.; Schwartz, S.D. A Proposed Mechanism for the Initial Myosin Binding Event on the Cardiac Thin Filament: A Metadynamics Study. J. Phys. Chem. Lett. 2021, 12, 3509–3513. [Google Scholar] [CrossRef]
- Marchenko, M.A.; Nefedova, V.V.; Yampolskaya, D.S.; Kopylova, G.V.; Shchepkin, D.V.; Bershitsky, S.Y.; Koubassova, N.A.; Tsaturyan, A.K.; Levitsky, D.I.; Matyushenko, A.M. Impact of A134 and E218 Amino Acid Residues of Tropomyosin on Its Flexibility and Function. Int. J. Mol. Sci. 2020, 21, 8720. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.B.; Lataro, R.C.; Ferro, J.A.; Reinach, F.d.C. Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 1994, 269, 10461–10466. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Koubassova, N.A.; Shchepkin, D.V.; Kopylova, G.V.; Nabiev, S.R.; Nikitina, L.V.; Bershitsky, S.Y.; Levitsky, D.I.; Tsaturyan, A.K. The effects of cardiomyopathy-associated mutations in the head-to-tail overlap junction of α-tropomyosin on its properties and interaction with actin. Int. J. Biol. Macromol. 2019, 125, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, V.V.; Koubassova, N.A.; Borzova, V.A.; Kleymenov, S.Y.; Tsaturyan, A.K.; Matyushenko, A.M.; Levitsky, D.I. Tropomyosin pseudo-phosphorylation can rescue the effects of cardiomyopathy-associated mutations. Int. J. Biol. Macromol. 2021, 166, 424–434. [Google Scholar] [CrossRef]
- Margossian, S.S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982, 85 Pt B, 55–71. [Google Scholar] [CrossRef]
- Reiser, P.J.; Kline, W.O. Electrophoretic separation and quantitation of cardiac myosin heavy chain isoforms in eight mammalian species. Am. J. Physiol. 1998, 274 Pt 2, H1048–H1053. [Google Scholar] [CrossRef]
- Pardee, J.D.; Spudich, J.A. Purification of muscle actin. Methods Enzymol. 1982, 85 Pt B, 164–179. [Google Scholar] [CrossRef]
- Potter, J.D. Preparation of troponin and its subunits. Methods Enzymol. 1982, 85 Pt B, 241–263. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Kleymenov, S.Y.; Susorov, D.S.; Levitsky, D.I. Thermal unfolding of homodimers and heterodimers of different skeletal-muscle isoforms of tropomyosin. Biophys. Chem. 2018, 243, 1–7. [Google Scholar] [CrossRef]
- Mashanov, G.I.; Molloy, J.E. Automatic detection of single fluorophores in live cells. Biophys. J. 2007, 92, 2199–2211. [Google Scholar] [CrossRef]
- Brunet, N.M.; Chase, P.B.; Mihajlović, G.; Schoffstall, B. Ca2+-regulatory function of the inhibitory peptide region of cardiac troponin I is aided by the C-terminus of cardiac troponin T: Effects of familial hypertrophic cardiomyopathy mutations cTnI R145G and cTnT R278C, alone and in combination, on filament sliding. Arch. Biochem. Biophys. 2014, 552–553, 11–20. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lindor-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber 99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tpm | Tmb (°C) | ΔHcal (kJ mol−1) | ΔHcal (% of Total) | Total ΔHcal c (kJ mol−1) |
|---|---|---|---|---|
| Tpm WT | 1330 | |||
| Domain 1 | 36.2 | 140 | 11 | |
| Domain 2 | 43.2 | 645 | 48 | |
| Domain 3 | 50.8 | 545 | 41 | |
| D219V Tpm | 1405 | |||
| Domain 1 | 41.9 | 390 | 28 | |
| Domain 2 | 45.7 | 630 | 45 | |
| Domain 3 | 51.7 | 385 | 27 |
| Myosin | Filament | Vmax (µm/s) | C50 (µg/mL) |
|---|---|---|---|
| Ventricular myosin | F-actin alone | 2.88 ± 0.01 | 20.1 ± 1.2 |
| WT Tpm | 1.50 ± 0.01 * | 54.9 ± 2.1 # | |
| D219V Tpm | 2.93 ± 0.01 | 26.0 ± 1.0 *, # | |
| Atrial myosin | F-actin alone | 3.26 ± 0.03 | 18.0 ± 1.4 |
| WT Tpm | 1.25 ± 0.01 * | 55.4 ± 0.2 # | |
| D219V Tpm | 3.32 ± 0.03 | 43.0 ± 0.1 *, # |
| Myosin | Tpm | Vmax (µm/s) | V0 (µm/s) | pCa50 |
|---|---|---|---|---|
| LV | WT Tpm | 3.73 ± 0.13 | 0.24 ± 0.06 | 6.22 ± 0.03 |
| D219V Tpm | 4.22 ± 0.05 | 0.81 ± 0.05 * | 6.65 ± 0.02 * | |
| LA | WT Tpm | 7.17 ± 0.10 | 0.39 ± 0.17 | 5.97 ± 0.03 |
| D219V Tpm | 8.45 ± 0.05 * | 1.42 ± 0.01 * | 6.27 ± 0.01 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsaturyan, A.K.; Zaklyazminskaya, E.V.; Polyak, M.E.; Kopylova, G.V.; Shchepkin, D.V.; Kochurova, A.M.; Gonchar, A.D.; Kleymenov, S.Y.; Koubasova, N.A.; Bershitsky, S.Y.; et al. De Novo Asp219Val Mutation in Cardiac Tropomyosin Associated with Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 18. https://doi.org/10.3390/ijms24010018
Tsaturyan AK, Zaklyazminskaya EV, Polyak ME, Kopylova GV, Shchepkin DV, Kochurova AM, Gonchar AD, Kleymenov SY, Koubasova NA, Bershitsky SY, et al. De Novo Asp219Val Mutation in Cardiac Tropomyosin Associated with Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences. 2023; 24(1):18. https://doi.org/10.3390/ijms24010018
Chicago/Turabian StyleTsaturyan, Andrey K., Elena V. Zaklyazminskaya, Margarita E. Polyak, Galina V. Kopylova, Daniil V. Shchepkin, Anastasia M. Kochurova, Anastasiia D. Gonchar, Sergey Y. Kleymenov, Natalia A. Koubasova, Sergey Y. Bershitsky, and et al. 2023. "De Novo Asp219Val Mutation in Cardiac Tropomyosin Associated with Hypertrophic Cardiomyopathy" International Journal of Molecular Sciences 24, no. 1: 18. https://doi.org/10.3390/ijms24010018
APA StyleTsaturyan, A. K., Zaklyazminskaya, E. V., Polyak, M. E., Kopylova, G. V., Shchepkin, D. V., Kochurova, A. M., Gonchar, A. D., Kleymenov, S. Y., Koubasova, N. A., Bershitsky, S. Y., Matyushenko, A. M., & Levitsky, D. I. (2023). De Novo Asp219Val Mutation in Cardiac Tropomyosin Associated with Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences, 24(1), 18. https://doi.org/10.3390/ijms24010018