
Fluorescence Lifetime Phasor Analysis of the Decamer–Dimer Equilibrium of Human Peroxiredoxin 1

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials
2.1. Human Prx1
2.2. Size Exclusion Chromatography
2.3. Fluorescence Emission Spectra
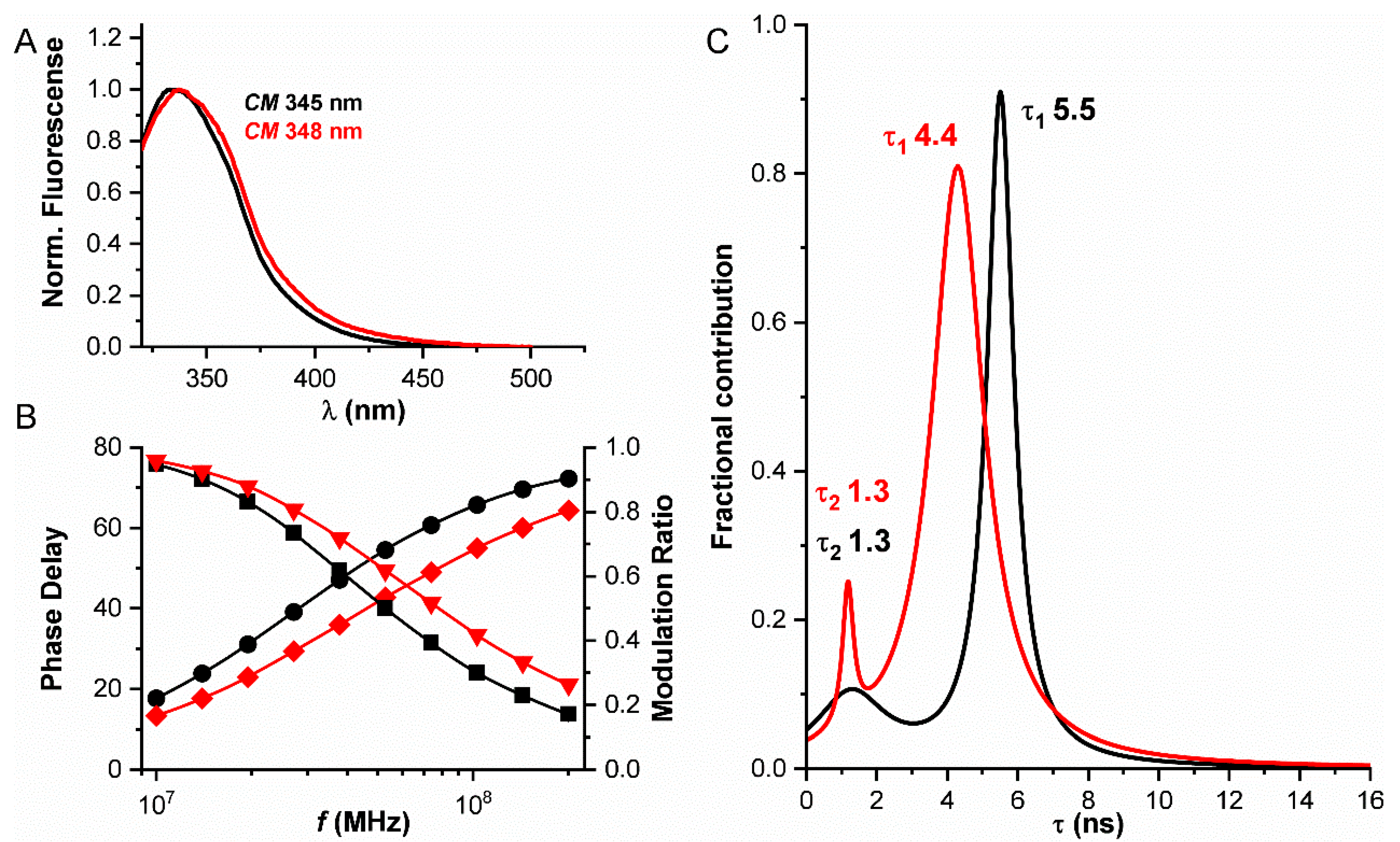
2.4. Fluorescence Lifetime Analog Frequency Domain Measurements
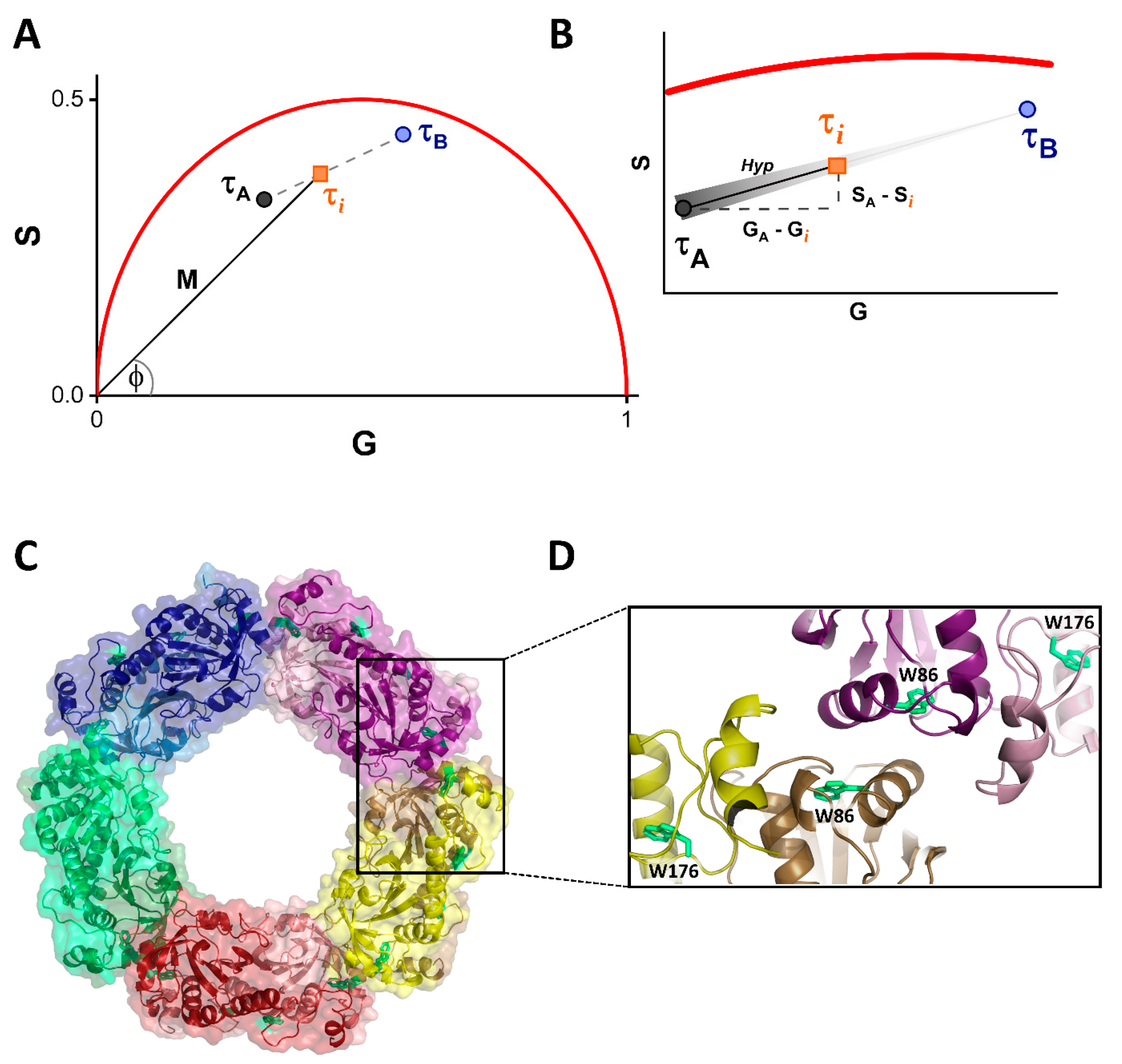
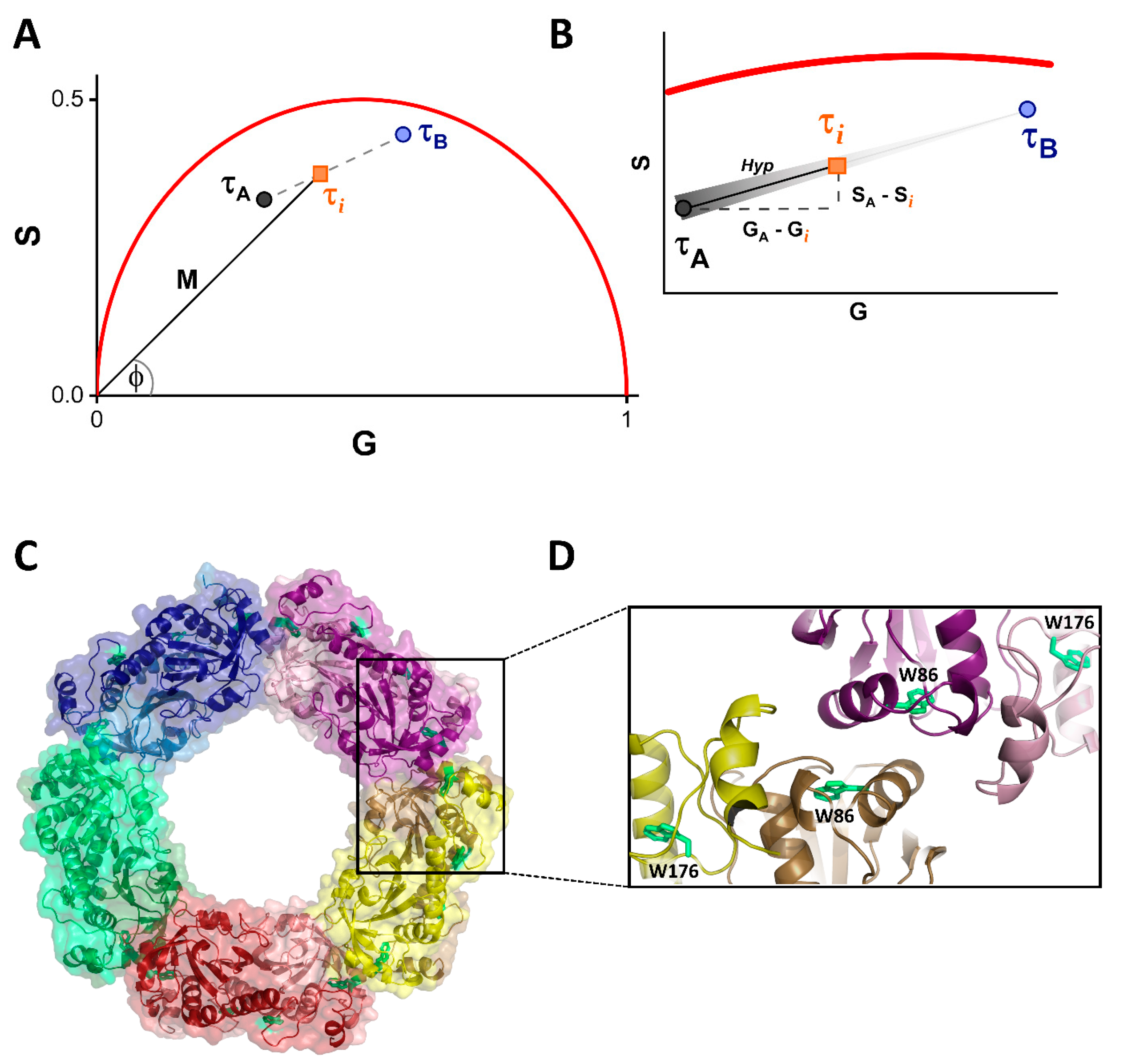
2.5. Fluorescence Lifetime Phasor Plot Analysis
2.6. Fitting Model
3. Results
3.1. Fluorescence Properties of hPrx1 and Their Dependance on Protein Concentration
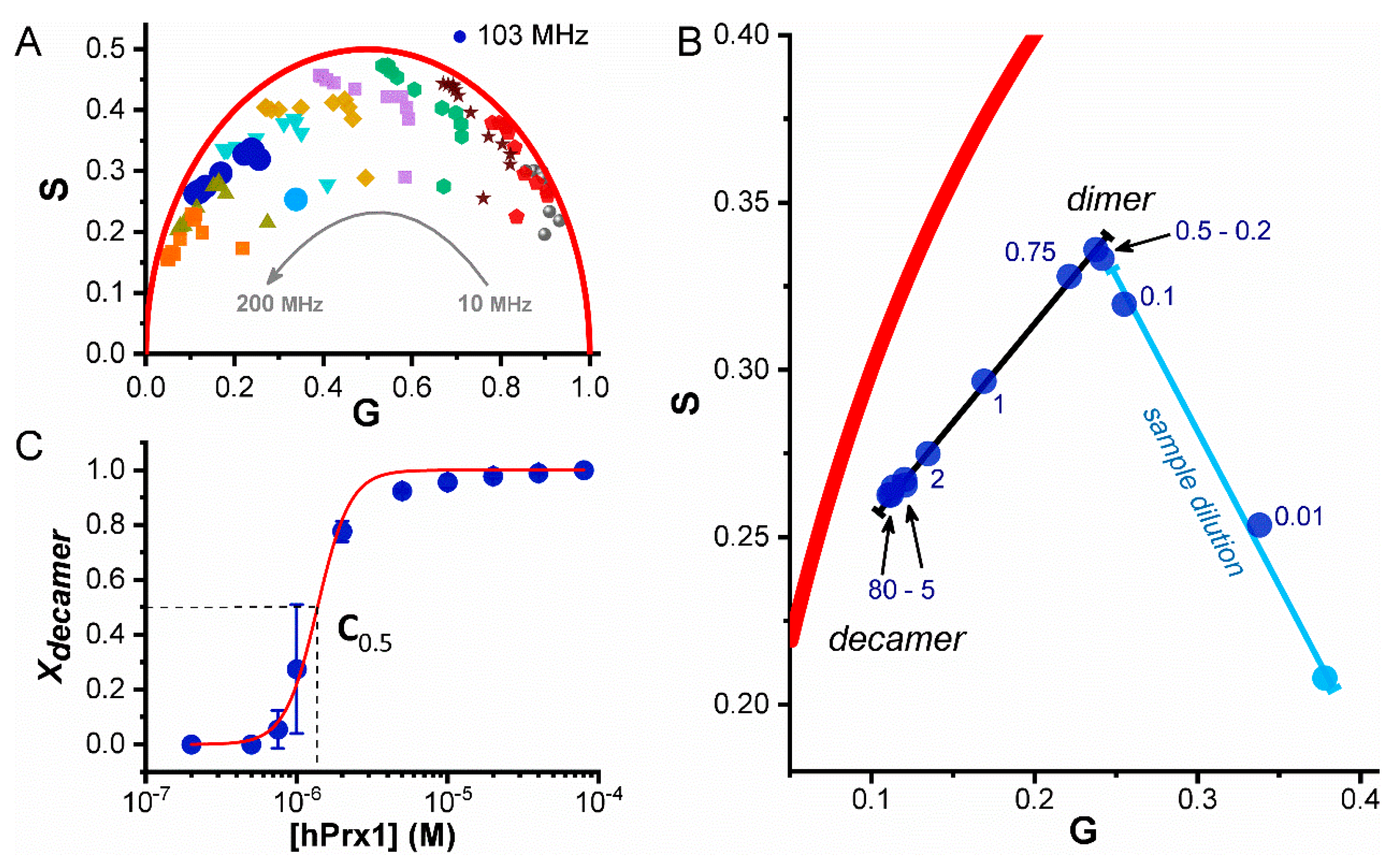
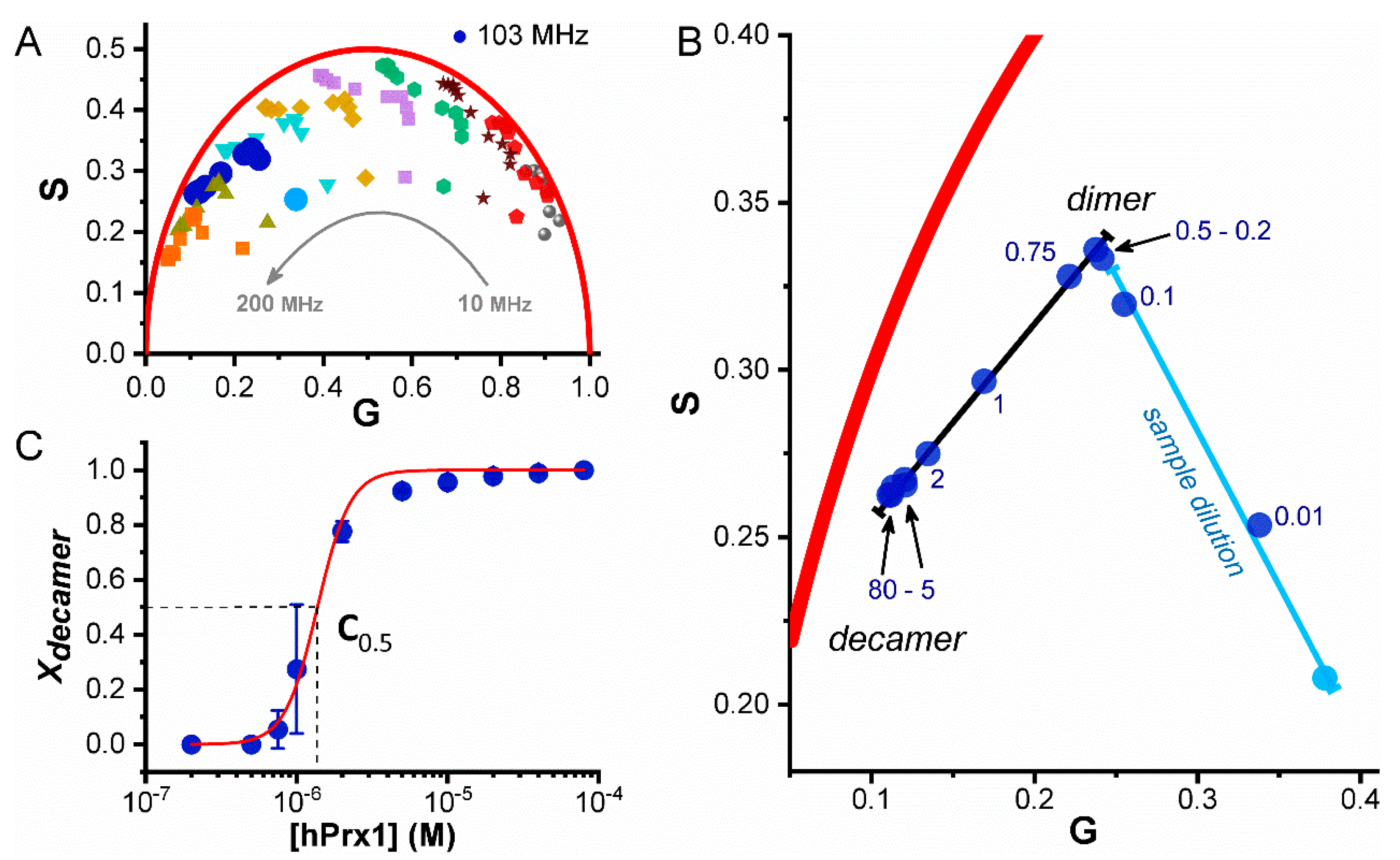
3.2. Dissociation of hPrx1 Followed by Fluorescence Lifetime Phasors
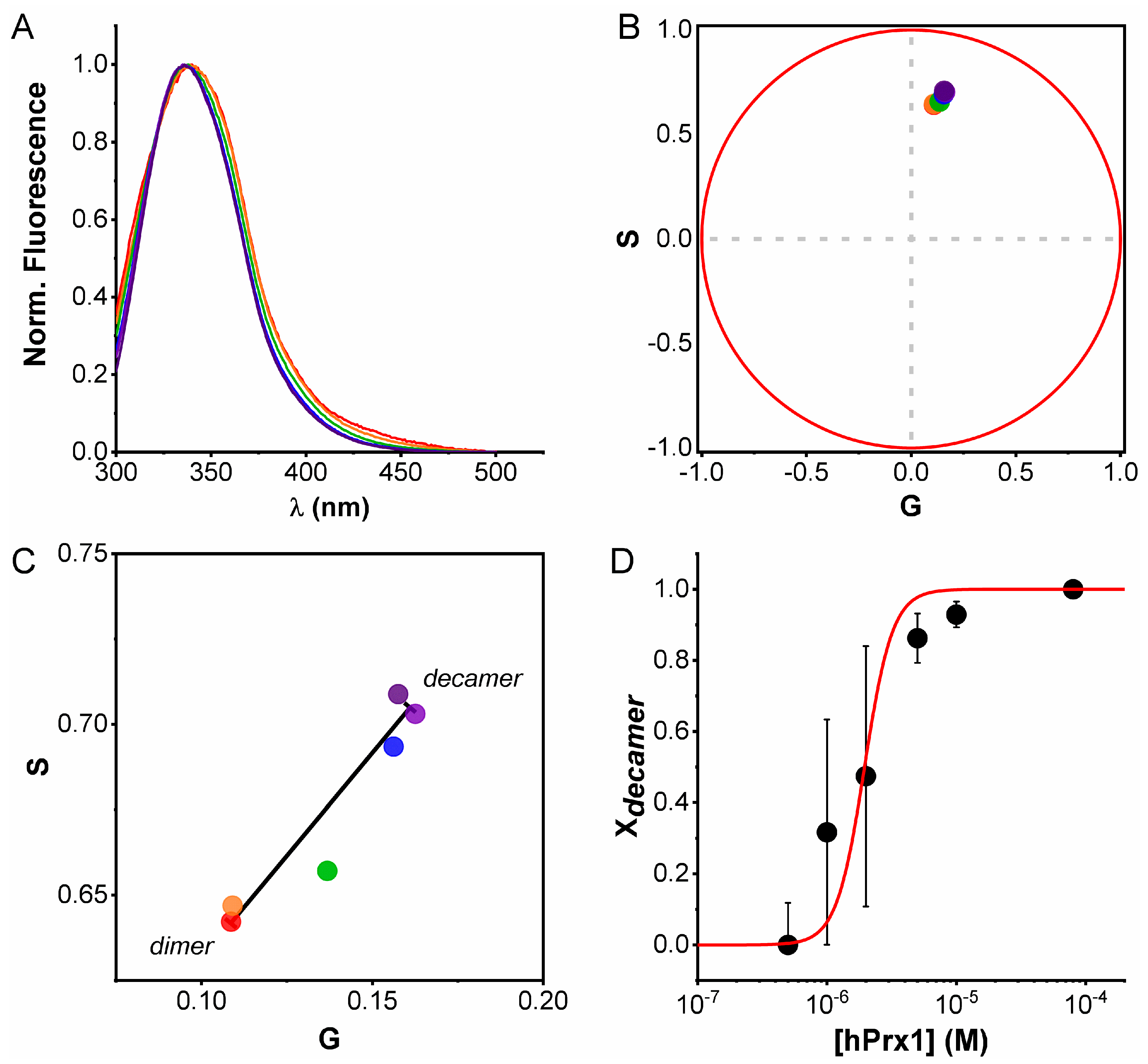
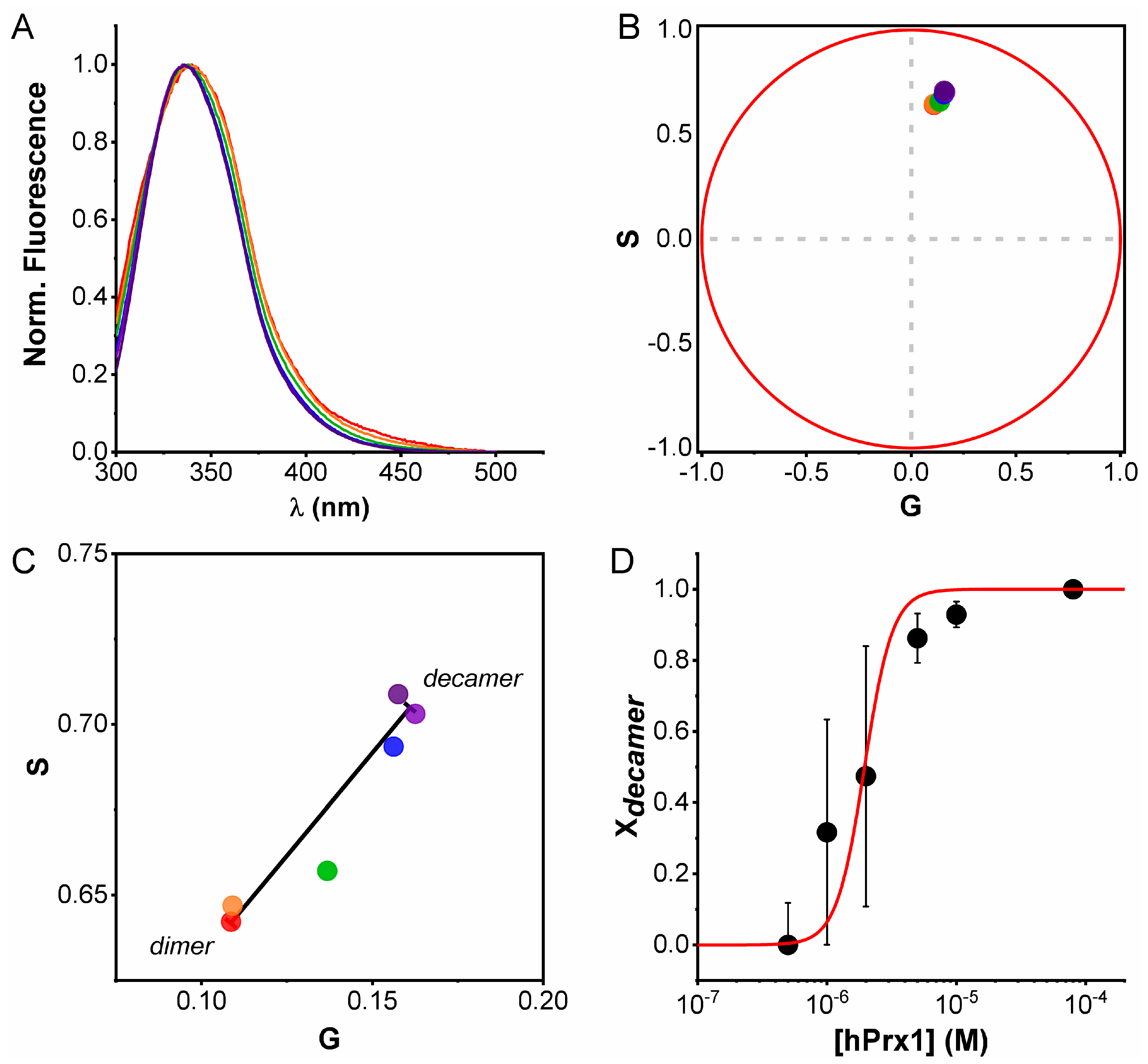
3.3. Emission Spectra Analysis Using Spectral Phasors
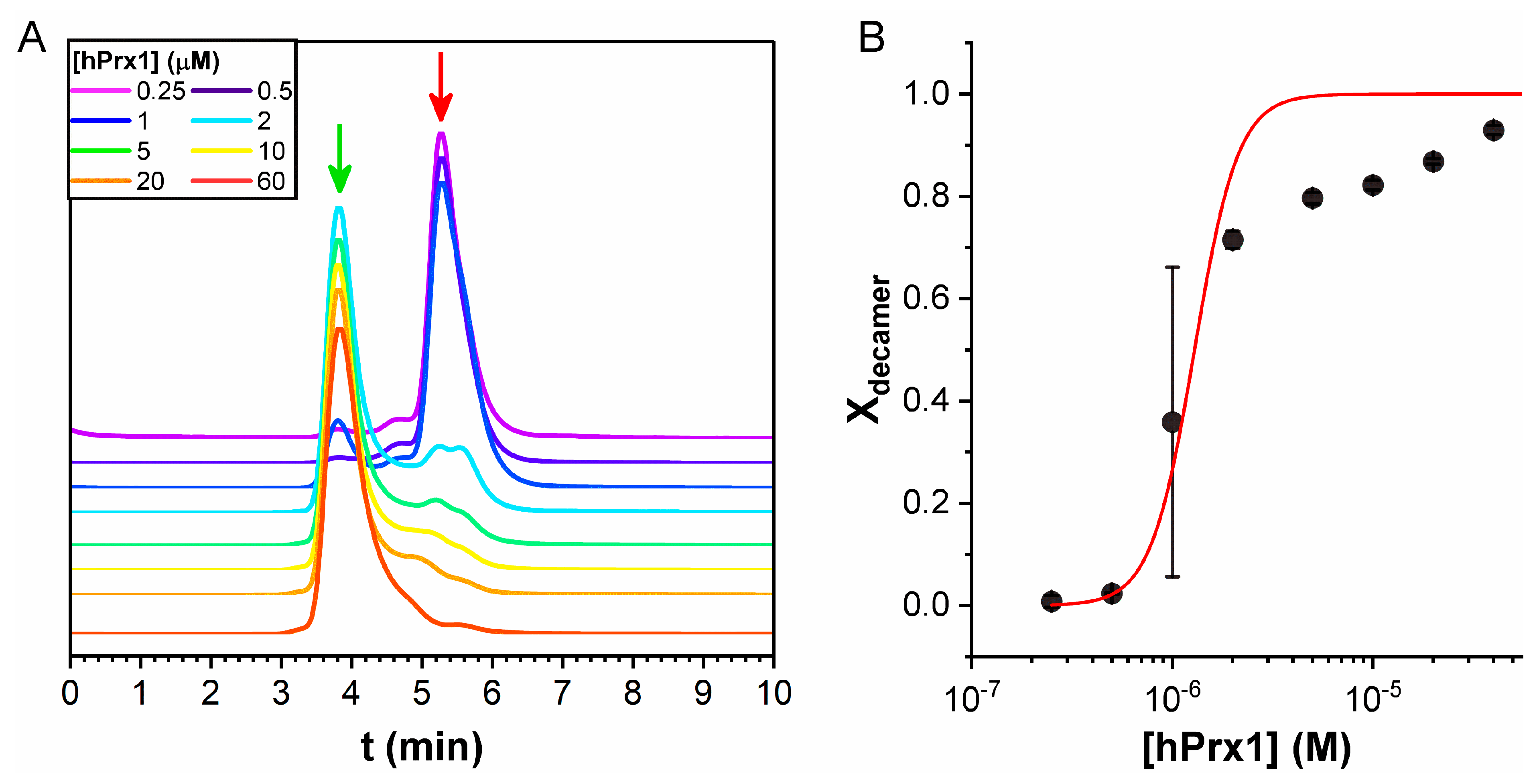
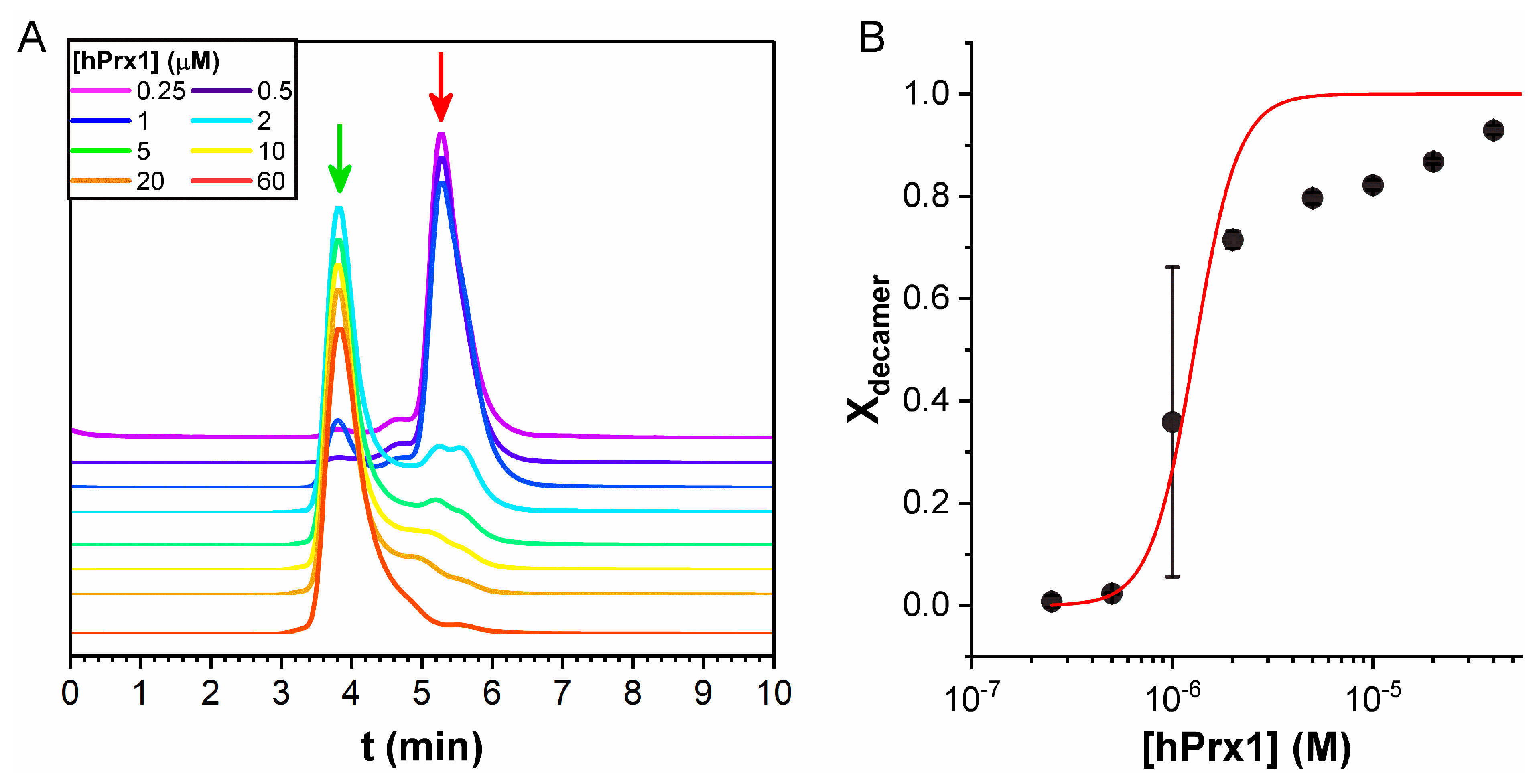
3.4. Size Exclusion Chromatography Analysis
4. Discussion
5. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Danielli, L.; Li, X.; Tuller, T.; Daniel, R. Quantifying the distribution of protein oligomerization degree reflects cellular information capacity. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Sunde, M. Dimers, Oligomers, Everywhere. Adv. Exp. Med. Biol. 2012, 747, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gell, D.A.; Grant, R.P.; Mackay, J.P. The Detection and Quantitation of Protein Oligomerization. Adv. Exp. Med. Biol. 2012, 747, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.J.; Dick, T.P. Fluorescent Protein-Based Redox Probes. Antioxid. Redox Signal. 2010, 13, 621–650. [Google Scholar] [CrossRef]
- Chen, Y.; Barkley, M.D. Toward Understanding Tryptophan Fluorescence in Proteins. Biochemistry 1998, 37, 9976–9982. [Google Scholar] [CrossRef]
- Ross, J.A.; Jameson, D.M. Time-resolved methods in biophysics. 8. Frequency domain fluorometry: Applications to intrinsic protein fluorescence. Photochem. Photobiol. Sci. 2008, 7, 1301–1312. [Google Scholar] [CrossRef]
- Gratton, E.; Silva, N.; Mei, G.; Rosato, N.; Savini, I.; Finazzi-Agro, A. Fluorescence lifetime distribution of folded and unfolded proteins. Int. J. Quantum Chem. 1992, 42, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Beechem, J.M.; Brand, L. Time-resolved fluorescence of proteins. Annu. Rev. Biochem. 1985, 54, 43–71. [Google Scholar] [CrossRef]
- Alcala, J.; Gratton, E.; Prendergast, F. Interpretation of fluorescence decays in proteins using continuous lifetime distributions. Biophys. J. 1987, 51, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, R.; Krishnamoorthy, G.; Periasamy, N. Similarity of fluorescence lifetime distributions for single tryptophan proteins in the random coil state. Biophys. J. 1994, 67, 2013–2023. [Google Scholar] [CrossRef] [Green Version]
- Malacrida, L.; Ranjit, S.; Jameson, D.M.; Gratton, E. The Phasor Plot: A Universal Circle to Advance Fluorescence Lifetime Analysis and Interpretation. Annu. Rev. Biophys. 2021, 50, 575–593. [Google Scholar] [CrossRef] [PubMed]
- Jameson, D.M. Introduction to Fluorescence; CRC Press: Boca Raton, FL, USA, 2014; pp. 1–286. [Google Scholar]
- Ranjit, S.; Malacrida, L.; Jameson, D.M.; Gratton, E. Fit-free analysis of fluorescence lifetime imaging data using the phasor approach. Nat. Protoc. 2018, 13, 1979–2004. [Google Scholar] [CrossRef] [PubMed]
- Štefl, M.; James, N.G.; Ross, J.A.; Jameson, D.M. Applications of phasors to in vitro time-resolved fluorescence measurements. Anal. Biochem. 2011, 410, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecinos-Franjola, F.; James, N.G.; Concha-Marambio, L.; Brunet, J.E.; Lagos, R.; Monasterio, O.; Jameson, D.M. Single tryptophan mutants of FtsZ: Nucleotide binding/exchange and conformational transitions. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1193–1200. [Google Scholar] [CrossRef]
- Lopes, J.L.S.; Araujo, A.P.U.; Jameson, D.M. Investigation of the conformational flexibility of DGAT1 peptides using tryptophan fluorescence. Methods Appl. Fluoresc. 2015, 3, 025003. [Google Scholar] [CrossRef] [Green Version]
- Weber, G. Resolution of the fluorescence lifetimes in a heterogeneous system by phase and modulation measurements. J. Phys. Chem. 1981, 85, 949–953. [Google Scholar] [CrossRef]
- Dalla-Rizza, J.; Randall, L.M.; Santos, J.; Ferrer-Sueta, G.; Denicola, A. Differential parameters between cytosolic 2-Cys peroxiredoxins, PRDX1 and PRDX2. Protein Sci. 2018, 28, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; Denicola, A.; Estrin, D.A.; Radi, R. Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols. Chem. Rev. 2019, 119, 10829–10855. [Google Scholar] [CrossRef]
- Troussicot, L.; Burmann, B.M.; Molin, M. Structural determinants of multimerization and dissociation in 2-Cys peroxiredoxin chaperone function. Structure 2021, 29, 640–654. [Google Scholar] [CrossRef]
- Lim, J.C.; Choi, H.-I.; Park, Y.S.; Nam, H.W.; Woo, H.A.; Kwon, K.-S.; Kim, Y.S.; Rhee, S.G.; Kim, K.; Chae, H.Z. Irreversible Oxidation of the Active-site Cysteine of Peroxiredoxin to Cysteine Sulfonic Acid for Enhanced Molecular Chaperone Activity. J. Biol. Chem. 2008, 283, 28873–28880. [Google Scholar] [CrossRef] [Green Version]
- Randall, L.M.; Ferrer-Sueta, G.; Denicola, A. Peroxiredoxins as Preferential Targets in H2O2-Induced Signaling. Methods Enzymol. 2013, 527, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stöcker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.D.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Ledesma, S.; Randall, L.M.; Parsonage, D.; Rizza, J.D.; Karplus, P.A.; Poole, L.; Denicola, A.; Ferrer-Sueta, G. Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry 2018, 57, 3416–3424. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Hecht, H.; Flohé, L. Peroxiredoxins. De Gruyter 2002, 383, 347–364. [Google Scholar] [CrossRef]
- Karplus, P.A. A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 2014, 80, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Wood, Z.A.; Poole, L.B.; Hantgan, R.R.; Karplus, P.A. Dimers to Doughnuts: Redox-Sensitive Oligomerization of 2-Cysteine Peroxiredoxins. Biochemistry 2002, 41, 5493–5504. [Google Scholar] [CrossRef]
- Radjainia, M.; Venugopal, H.; Desfosses, A.; Phillips, A.J.; Yewdall, N.A.; Hampton, M.B.; Gerrard, J.A.; Mitra, A.K. Cryo-Electron Microscopy Structure of Human Peroxiredoxin-3 Filament Reveals the Assembly of a Putative Chaperone. Structure 2015, 23, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Conroy, F.; Rossi, T.; Ashmead, H.; Crowther, J.M.; Mitra, A.K.; Gerrard, J.A. Engineering peroxiredoxin 3 to facilitate control over self-assembly. Biochem. Biophys. Res. Commun. 2019, 512, 263–268. [Google Scholar] [CrossRef]
- Socas, L.B.P.; Ambroggio, E.E. The influence of myristoylation, liposome surface charge and nucleic acid interaction in the partition properties of HIV-1 Gag-N-terminal peptides to membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183421. [Google Scholar] [CrossRef]
- Malacrida, L.; Gratton, E.; Jameson, D.M. Model-free methods to study membrane environmental probes: A comparison of the spectral phasor and generalized polarization approaches. Methods Appl. Fluoresc. 2015, 3. [Google Scholar] [CrossRef]
- James, N.G.; Ross, J.A.; Štefl, M.; Jameson, D.M. Applications of phasor plots to in vitro protein studies. Anal. Biochem. 2011, 410, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tairum, C.A.; de Oliveira, M.A.; Horta, B.B.; Zara, F.J.; Netto, L.E. Disulfide Biochemistry in 2-Cys Peroxiredoxin: Requirement of Glu50 and Arg146 for the Reduction of Yeast Tsa1 by Thioredoxin. J. Mol. Biol. 2012, 424, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Schröder, E.; Willis, A.C.; Ponting, C.P. Porcine natural-killer-enhancing factor-B: Oligomerisation and identification as a calpain substrate in vitro. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1998, 1383, 279–291. [Google Scholar] [CrossRef]
- Cao, Z.; Bhella, D.; Lindsay, J.G. Reconstitution of the Mitochondrial PrxIII Antioxidant Defence Pathway: General Properties and Factors Affecting PrxIII Activity and Oligomeric State. J. Mol. Biol. 2007, 372, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Plishker, G.; Chevalier, D.; Seinsoth, L.; Moore, R. Calcium-activated potassium transport and high molecular weight forms of calpromotin. J. Biol. Chem. 1992, 267, 21839–21843. [Google Scholar] [CrossRef]
- Himiyama, T.; Nakamura, T. Disassembly of the ring-type decameric structure of peroxiredoxin from Aeropyrum pernix K1 by amino acid mutation. Protein Sci. 2020, 29, 1138–1147. [Google Scholar] [CrossRef]
- Manta, B.; Hugo, M.; Ortiz, C.; Ferrer-Sueta, G.; Trujillo, M.; Denicola, A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 2009, 484, 146–154. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors Affecting Protein Thiol Reactivity and Specificity in Peroxide Reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef]
- Selvaggio, G.; Coelho, P.M.; Salvador, A. Mapping the phenotypic repertoire of the cytoplasmic 2-Cys peroxiredoxin—Thioredoxin system. 1. Understanding commonalities and differences among cell types. Redox Biol. 2017, 15, 297–315. [Google Scholar] [CrossRef]
- Barranco-Medina, S.; Kakorin, S.; Lázaro, J.J.; Dietz, K.-J. Thermodynamics of the Dimer−Decamer Transition of Reduced Human and Plant 2-Cys Peroxiredoxin. Biochemistry 2008, 47, 7196–7204. [Google Scholar] [CrossRef]
- Liebthal, M.; Kushwah, M.S.; Kukura, P.; Dietz, K.-J. Single molecule mass photometry reveals the dynamic oligomerization of human and plant peroxiredoxins. iScience 2021, 24, 103258. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villar, S.F.; Dalla-Rizza, J.; Möller, M.N.; Ferrer-Sueta, G.; Malacrida, L.; Jameson, D.M.; Denicola, A. Fluorescence Lifetime Phasor Analysis of the Decamer–Dimer Equilibrium of Human Peroxiredoxin 1. Int. J. Mol. Sci. 2022, 23, 5260. https://doi.org/10.3390/ijms23095260
Villar SF, Dalla-Rizza J, Möller MN, Ferrer-Sueta G, Malacrida L, Jameson DM, Denicola A. Fluorescence Lifetime Phasor Analysis of the Decamer–Dimer Equilibrium of Human Peroxiredoxin 1. International Journal of Molecular Sciences. 2022; 23(9):5260. https://doi.org/10.3390/ijms23095260
Chicago/Turabian StyleVillar, Sebastián F., Joaquín Dalla-Rizza, Matías N. Möller, Gerardo Ferrer-Sueta, Leonel Malacrida, David M. Jameson, and Ana Denicola. 2022. "Fluorescence Lifetime Phasor Analysis of the Decamer–Dimer Equilibrium of Human Peroxiredoxin 1" International Journal of Molecular Sciences 23, no. 9: 5260. https://doi.org/10.3390/ijms23095260
APA StyleVillar, S. F., Dalla-Rizza, J., Möller, M. N., Ferrer-Sueta, G., Malacrida, L., Jameson, D. M., & Denicola, A. (2022). Fluorescence Lifetime Phasor Analysis of the Decamer–Dimer Equilibrium of Human Peroxiredoxin 1. International Journal of Molecular Sciences, 23(9), 5260. https://doi.org/10.3390/ijms23095260