
Add and Go: FRET Acceptor for Live-Cell Measurements Modulated by Externally Provided Ligand


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
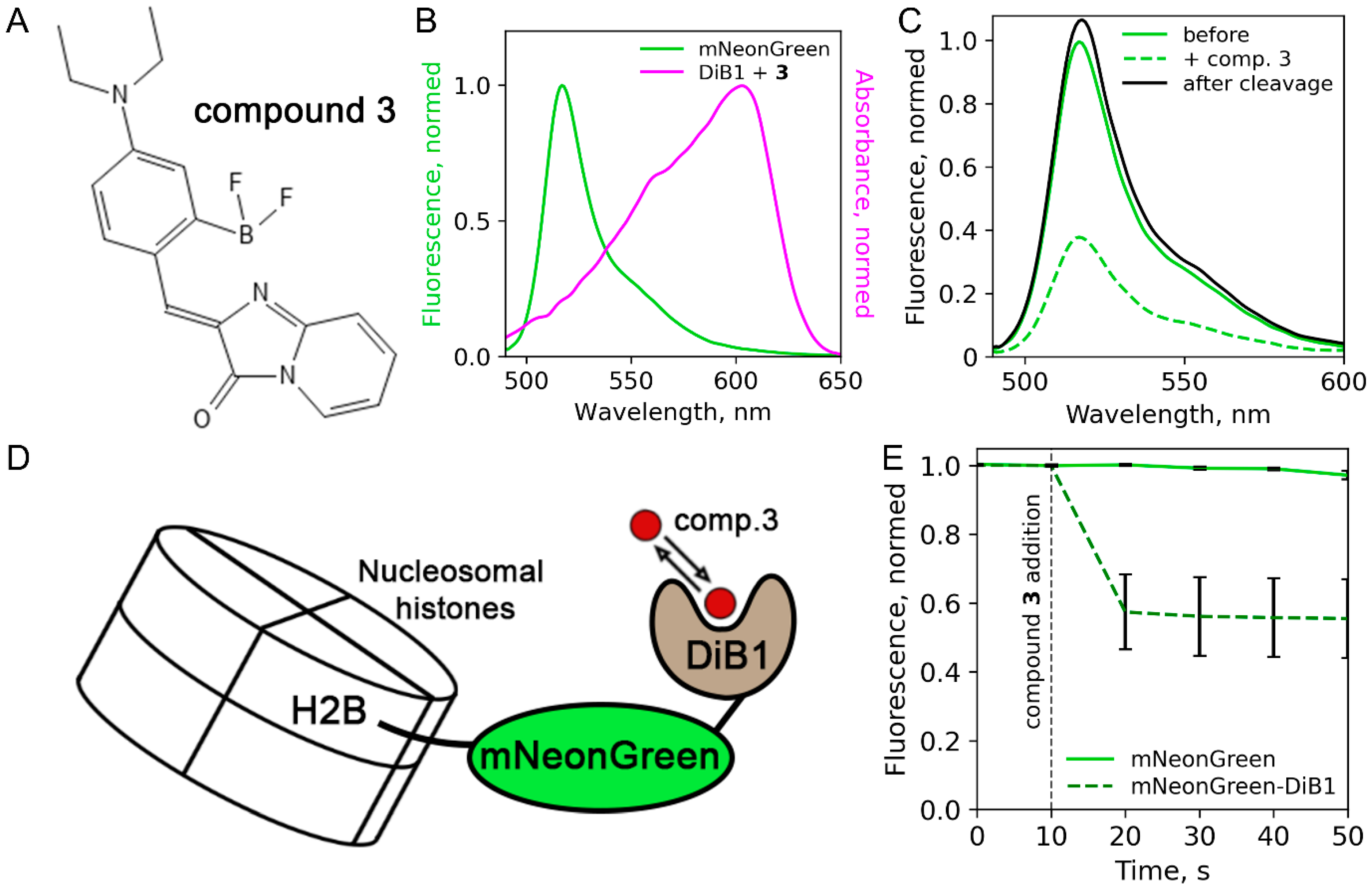
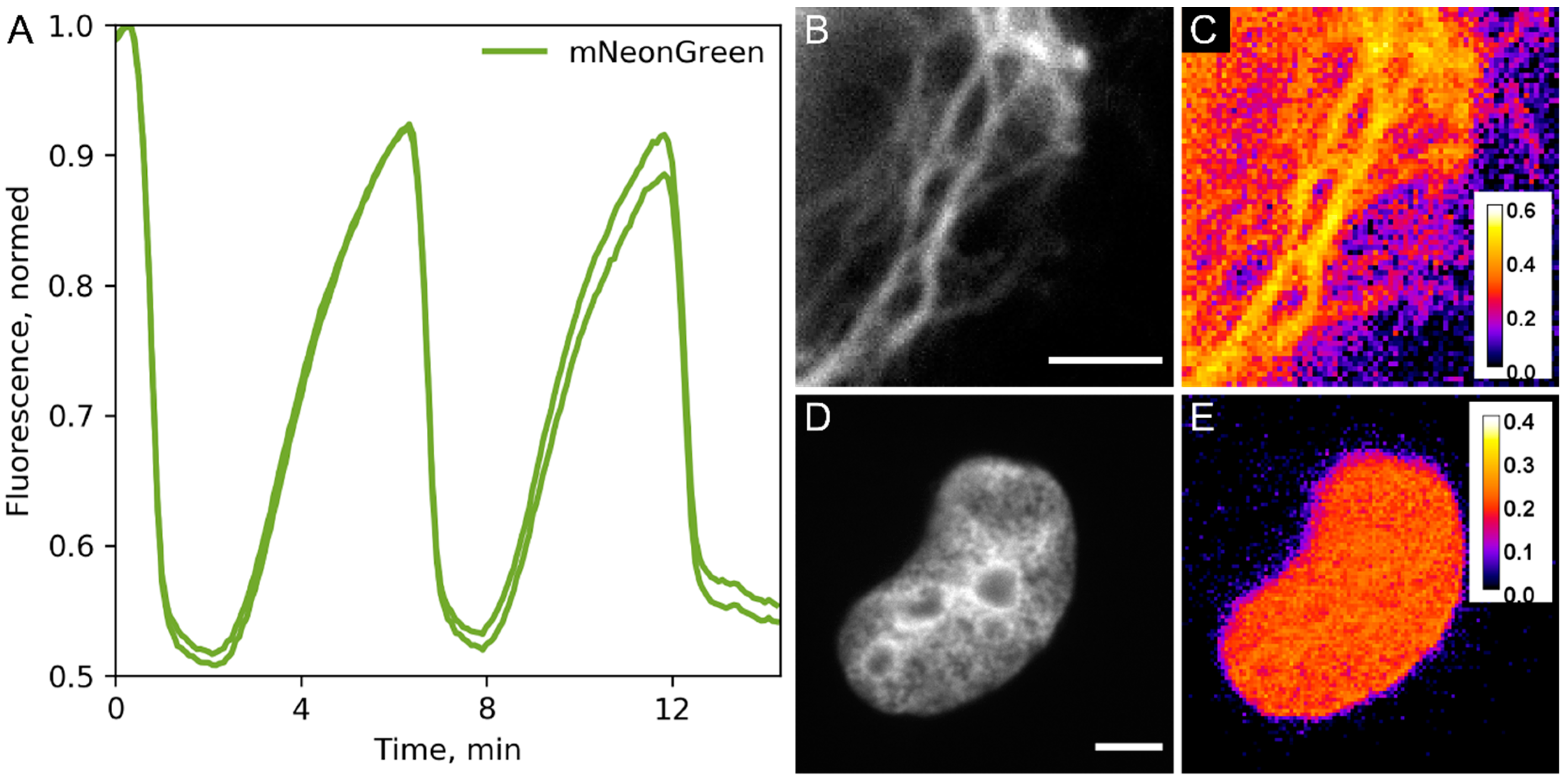
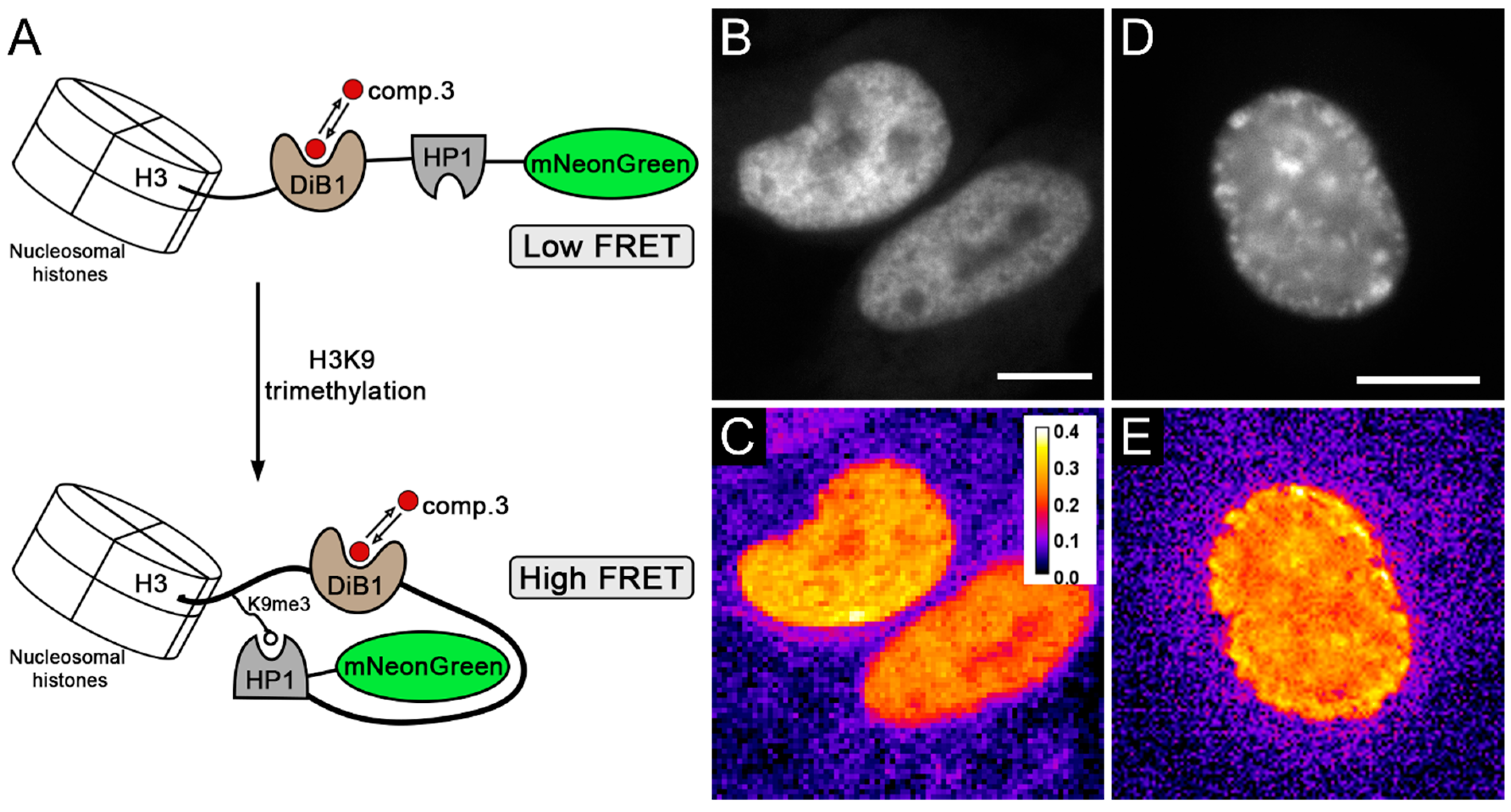
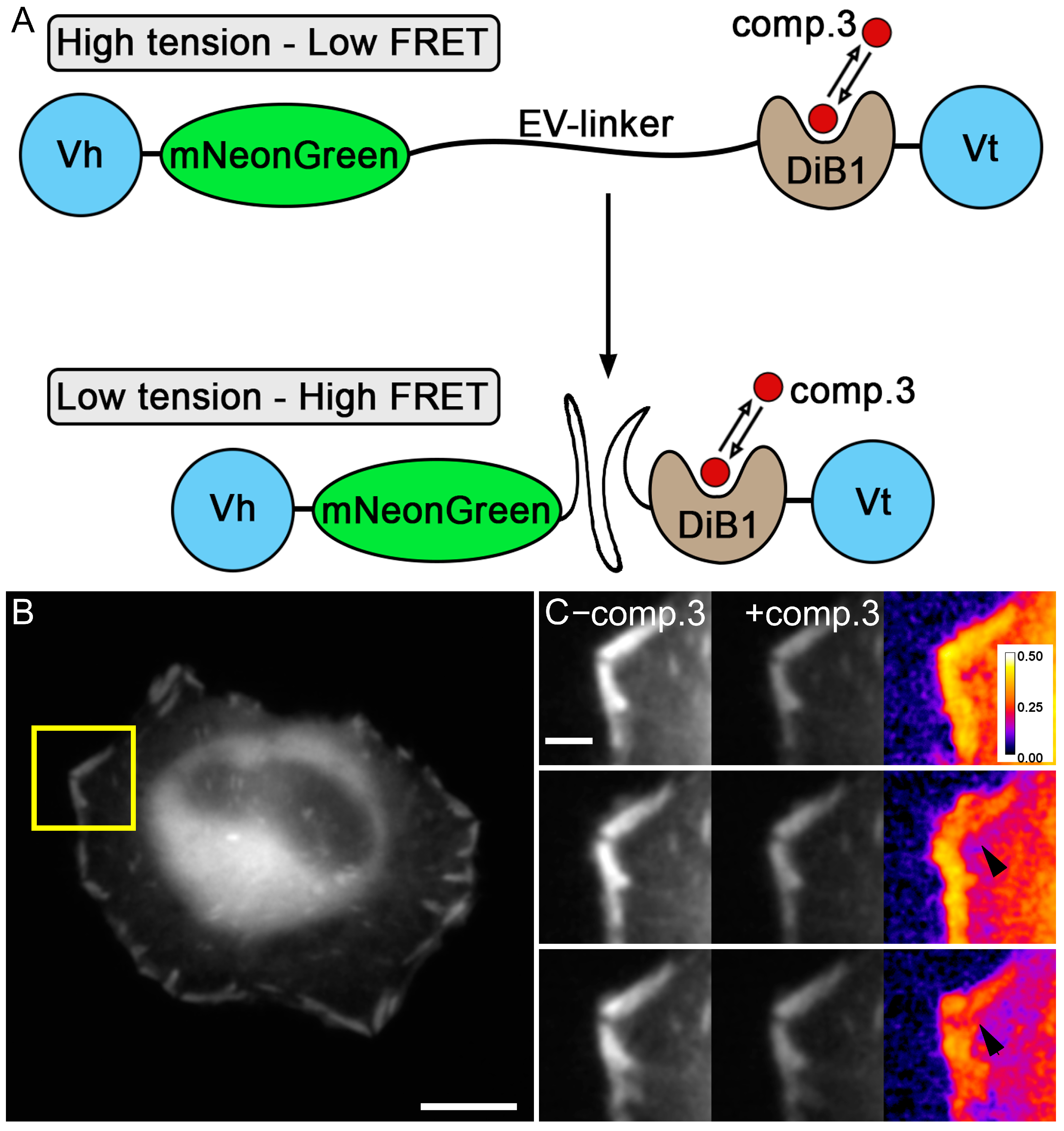
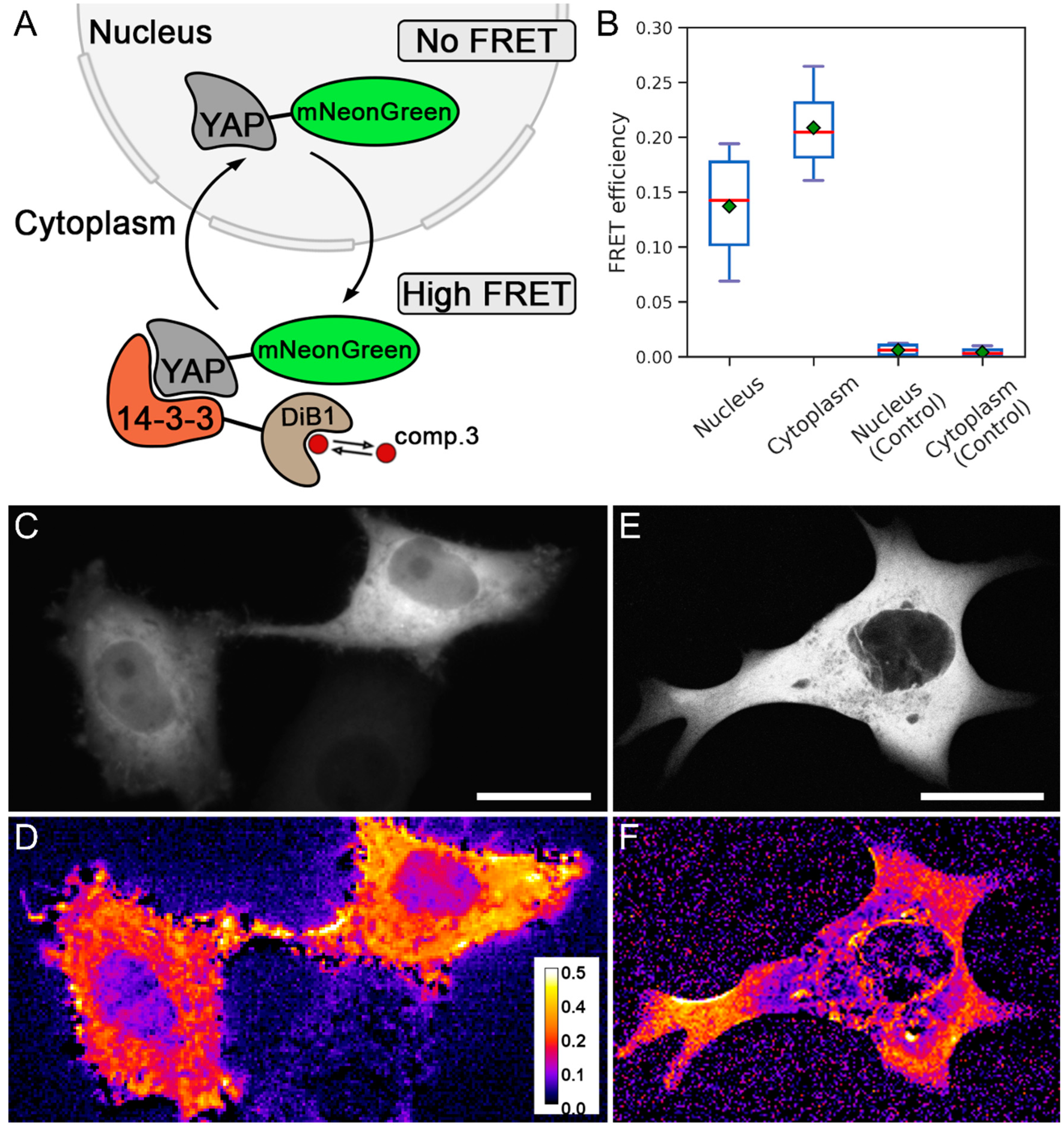
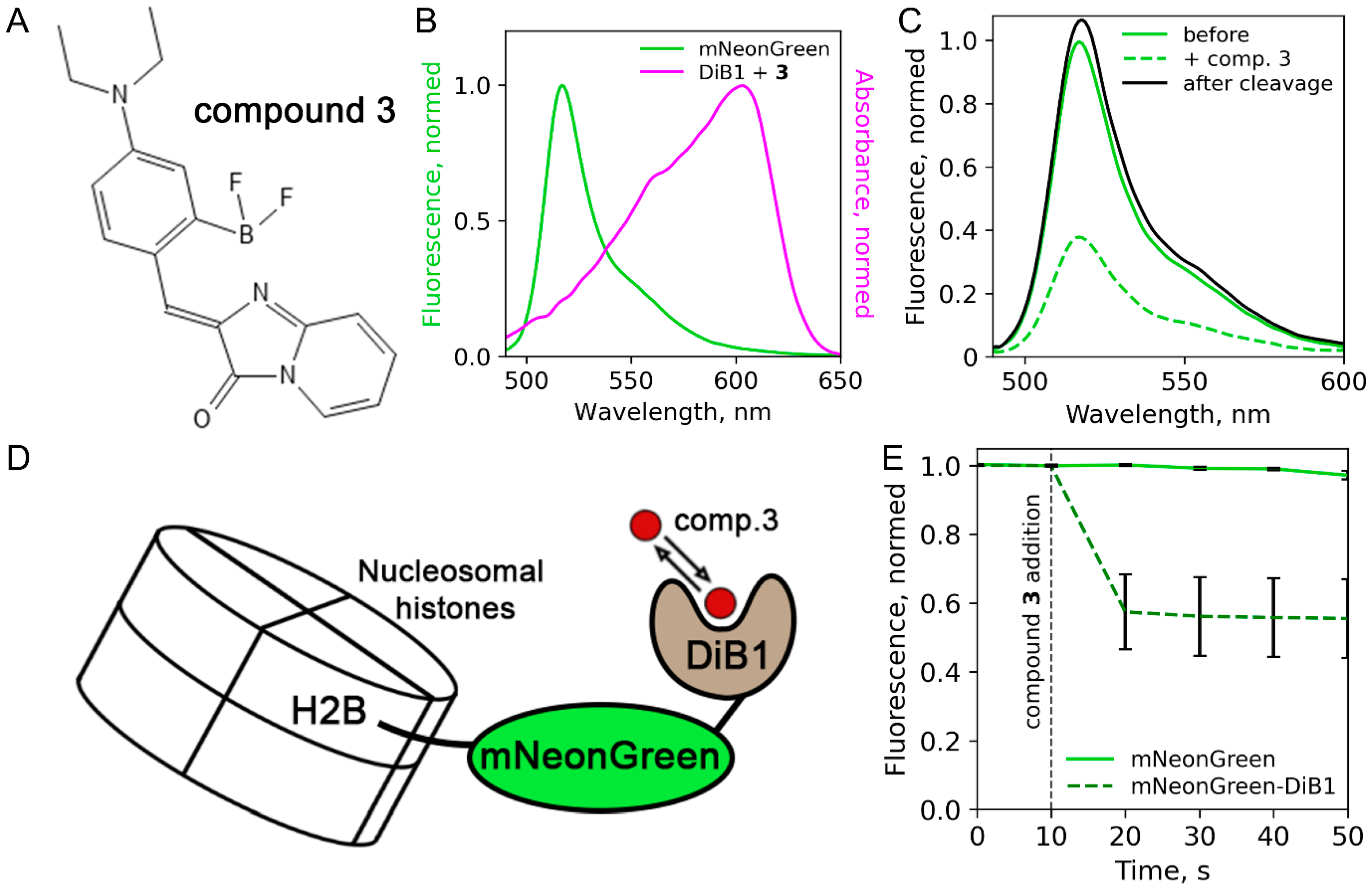
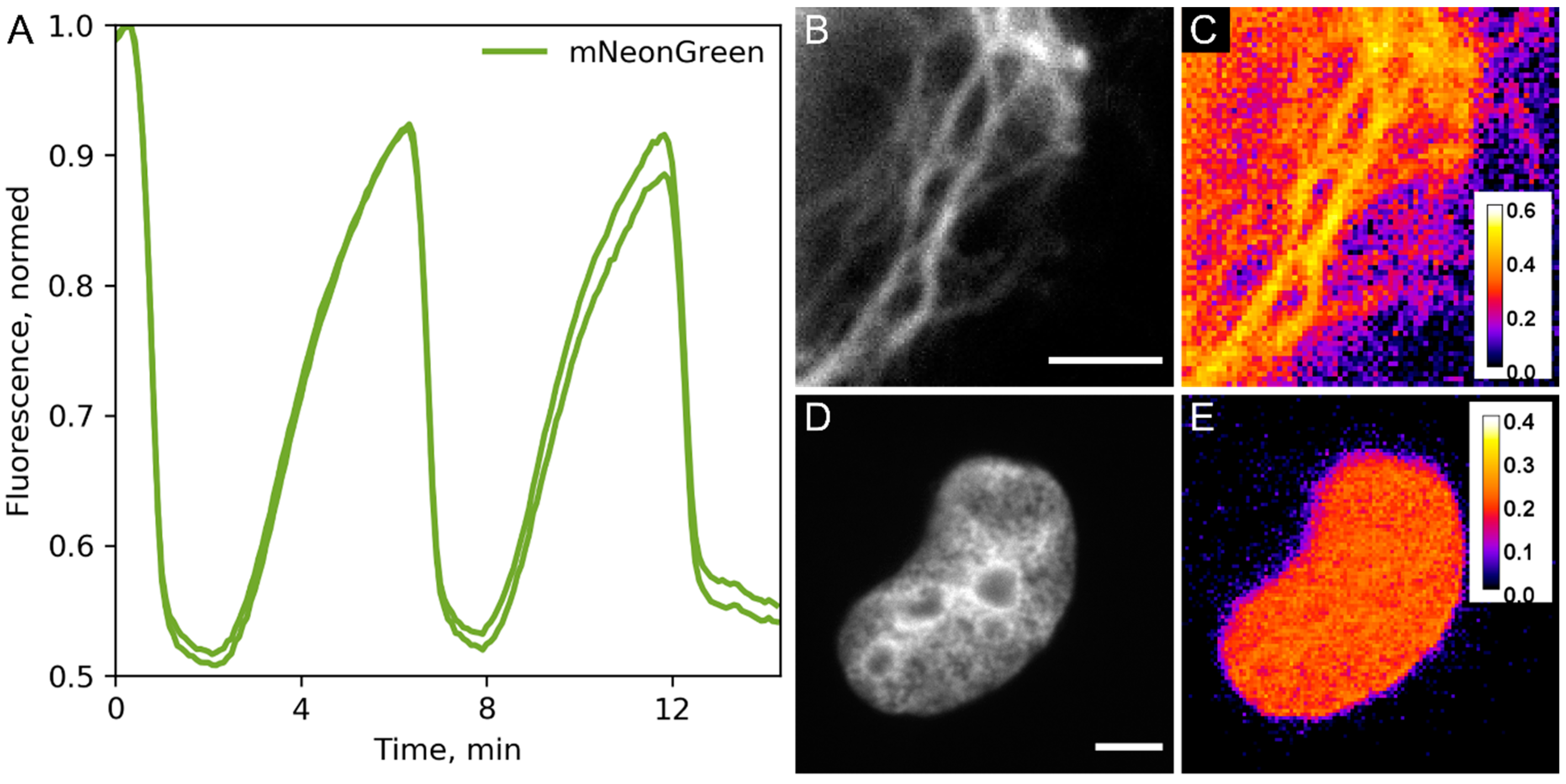
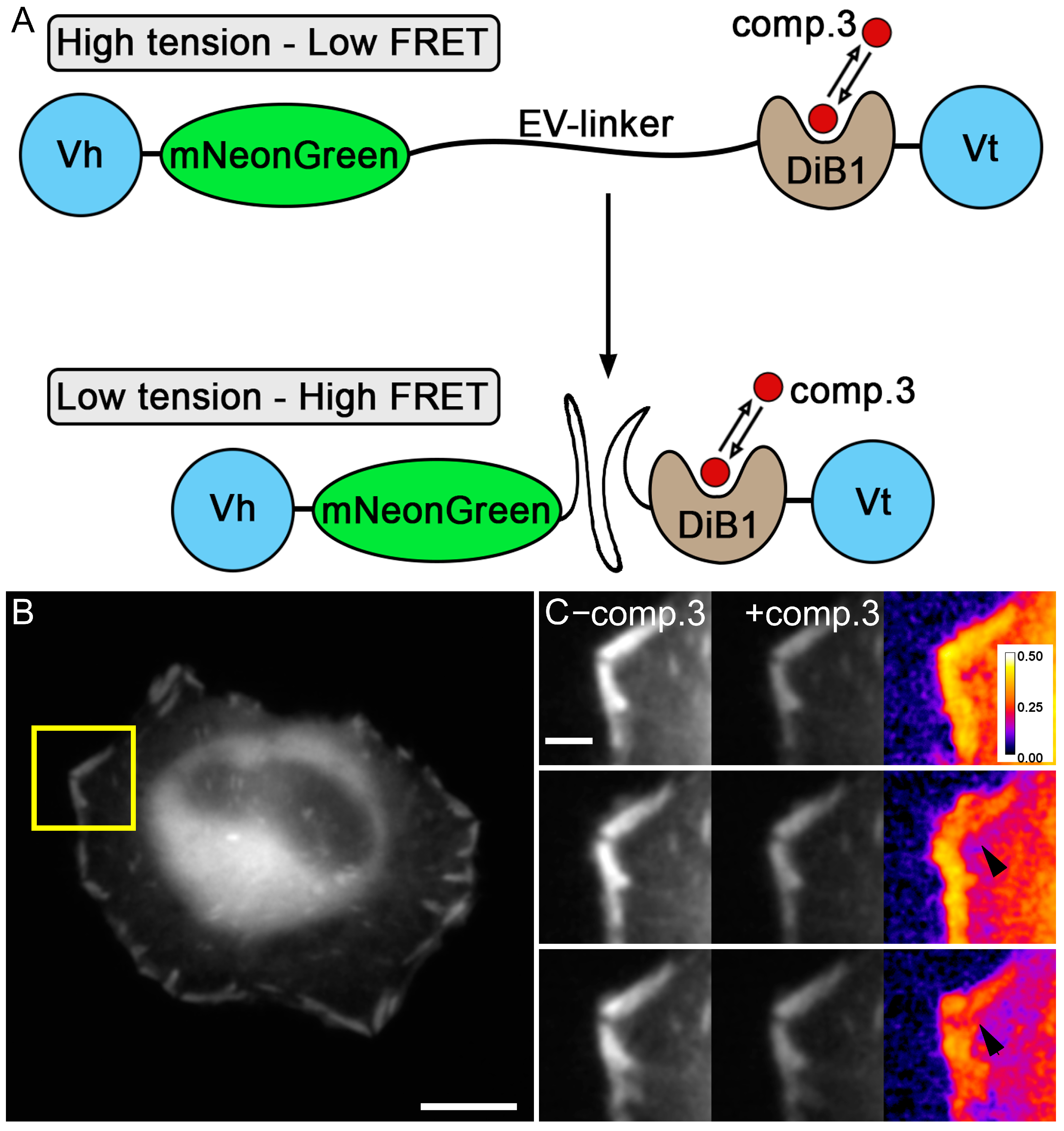
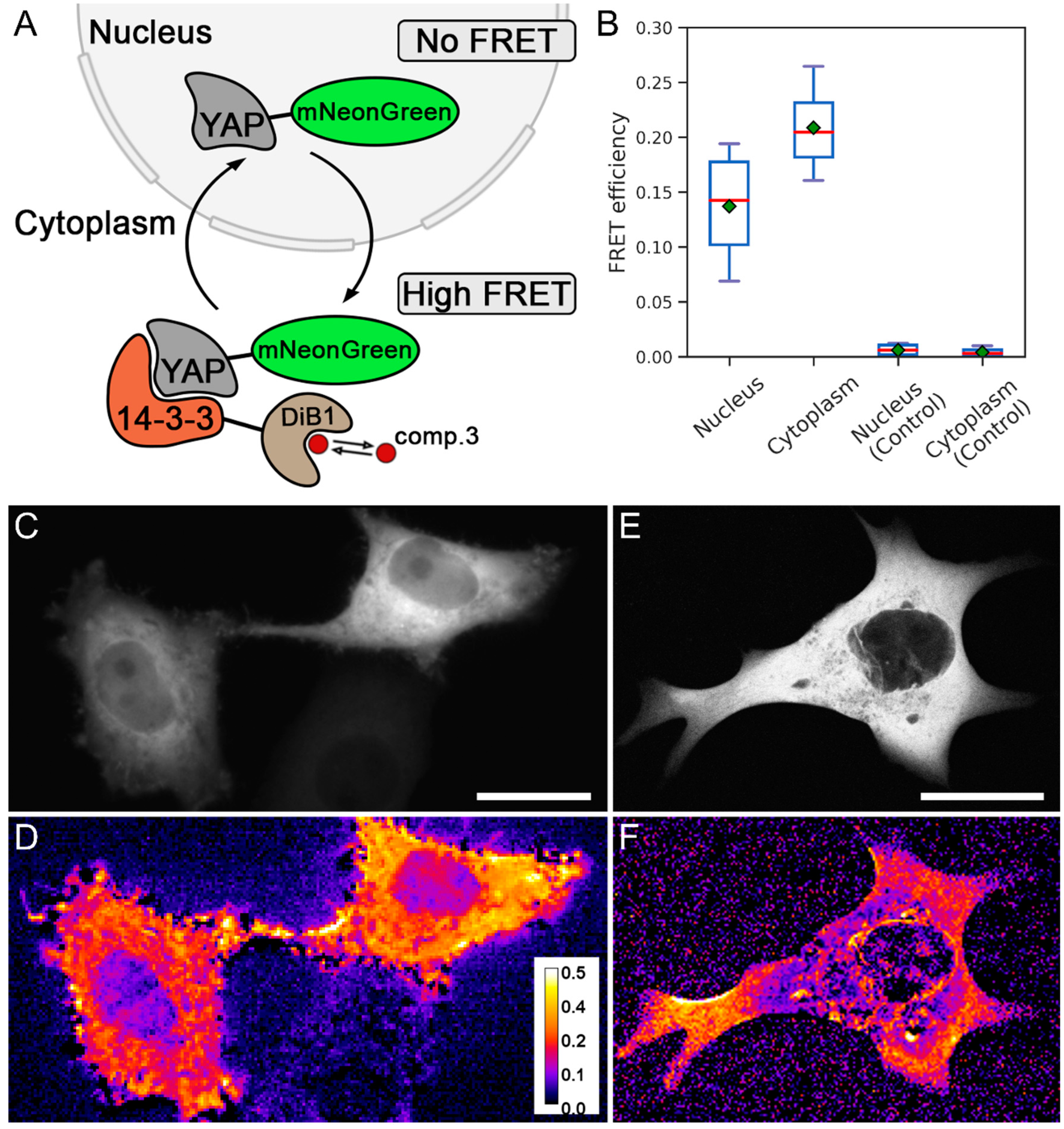
2. Results and Discussion
3. Materials and Methods
3.1. Molecular Cloning
3.2. Protein Expression and Purification
3.3. Fluorescence Spectra and Förster Radius Evaluation
3.4. Cell Culture and Transient Transfection
3.5. Cell Culture Fixation
3.6. Widefield Fluorescence Microscopy and Washing
3.7. Confocal Microscopy
3.8. FRET Efficiency Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Koldenkova, V.P.; Nagai, T. Genetically Encoded Ca2 Indicators: Properties and Evaluation. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Grashoff, C.; Hoffman, B.D.; Brenner, M.D.; Zhou, R.; Parsons, M.; Yang, M.T.; McLean, M.A.; Sligar, S.G.; Chen, C.S.; Ha, T.; et al. Measuring Mechanical Tension across Vinculin Reveals Regulation of Focal Adhesion Dynamics. Nature 2010, 466, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Russwurm, M.; Koesling, D. Measurement of cGMP-Generating and -Degrading Activities and cGMP Levels in Cells and Tissues: Focus on FRET-Based cGMP Indicators. Nitric Oxide 2018, 77, 44–52. [Google Scholar] [CrossRef]
- Rowland, C.E.; Brown, C.W.; Medintz, I.L.; Delehanty, J.B. Intracellular FRET-Based Probes: A Review. Methods Appl. Fluoresc. 2015, 3, 42006. [Google Scholar] [CrossRef]
- Van Munster, E.B.; Kremers, G.J.; Adjobo-Hermans, M.J.W.; Gadella, T.W.J., Jr. Fluorescence Resonance Energy Transfer (FRET) Measurement by Gradual Acceptor Photobleaching. J. Microsc. 2005, 218, 253–262. [Google Scholar] [CrossRef]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef]
- Subach, F.V.; Zhang, L.; Gadella, T.W.J.; Gurskaya, N.G.; Lukyanov, K.A.; Verkhusha, V.V. Red Fluorescent Protein with Reversibly Photoswitchable Absorbance for Photochromic FRET. Chem. Biol. 2010, 17, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Giordano, L.; Jovin, T.M.; Irie, M.; Jares-Erijman, E.A. Diheteroarylethenes as Thermally Stable Photoswitchable Acceptors in Photochromic Fluorescence Resonance Energy Transfer (pcFRET). J. Am. Chem. Soc. 2002, 124, 7481–7489. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, C.; Schmidt, B.F.; Schmidt, B.A.; Creeger, Y.; Fisher, G.W.; Zakel, K.L.; Adler, S.; Fitzpatrick, J.A.J.; Woolford, C.A.; Yan, Q.; et al. Fluorogen-Activating Single-Chain Antibodies for Imaging Cell Surface Proteins. Nat. Biotechnol. 2008, 26, 235–240. [Google Scholar] [CrossRef]
- Plamont, M.-A.; Billon-Denis, E.; Maurin, S.; Gauron, C.; Pimenta, F.M.; Specht, C.G.; Shi, J.; Quérard, J.; Pan, B.; Rossignol, J.; et al. Small Fluorescence-Activating and Absorption-Shifting Tag for Tunable Protein Imaging in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Bozhanova, N.G.; Baranov, M.S.; Klementieva, N.V.; Sarkisyan, K.S.; Gavrikov, A.S.; Yampolsky, I.V.; Zagaynova, E.V.; Lukyanov, S.A.; Lukyanov, K.A.; Mishin, A.S. Protein Labeling for Live Cell Fluorescence Microscopy with a Highly Photostable Renewable Signal. Chem. Sci. 2017, 8, 7138–7142. [Google Scholar] [CrossRef] [Green Version]
- Benaissa, H.; Ounoughi, K.; Aujard, I.; Fischer, E.; Goïame, R.; Nguyen, J.; Tebo, A.G.; Li, C.; Le Saux, T.; Bertolin, G.; et al. Engineering of a Fluorescent Chemogenetic Reporter with Tunable Color for Advanced Live-Cell Imaging. Nat. Commun. 2021, 12, 6989. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A Bright Monomeric Green Fluorescent Protein Derived from Branchiostoma Lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Bozhanova, N.G.; Baranov, M.S.; Baleeva, N.S.; Gavrikov, A.S.; Mishin, A.S. Red-Shifted Aminated Derivatives of GFP Chromophore for Live-Cell Protein Labeling with Lipocalins. Int. J. Mol. Sci. 2018, 19, 3778. [Google Scholar] [CrossRef] [Green Version]
- Kursunlu, A.N. Synthesis and Photophysical Properties of Modifiable Single, Dual, and Triple-Boron Dipyrromethene (Bodipy) Complexes. Tetrahedron Lett. 2015, 56, 1873–1877. [Google Scholar] [CrossRef]
- Kursunlu, A.N.; Baslak, C. A Bodipy-Bearing pillar [5] arene for Mimicking Photosynthesis: Multi-Fluorophoric Light Harvesting System. Tetrahedron Lett. 2018, 59, 1958–1962. [Google Scholar] [CrossRef]
- Kursunlu, A.N.; Oguz, M.; Yilmaz, M. On/Off Rhodamine-BODIPY-Based Fluorimetric/Colorimetric Sensor for Detection of Mercury (II) in Half-Aqueous Medium. IEEE Sens. J. 2019, 19, 2009–2015. [Google Scholar] [CrossRef]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving Brightness and Photostability of Green and Red Fluorescent Proteins for Live Cell Imaging and FRET Reporting. Sci. Rep. 2016, 6, 20889. [Google Scholar] [CrossRef]
- Gaume, X.; Monier, K.; Argoul, F.; Mongelard, F.; Bouvet, P. In Vivo Study of the Histone Chaperone Activity of Nucleolin by FRAP. Biochem. Res. Int. 2011, 2011, 187624. [Google Scholar] [CrossRef] [Green Version]
- Pang, A.H.; Obiero, J.M.; Kulczyk, A.W.; Sviripa, V.M.; Tsodikov, O.V. A Crystal Structure of Coil 1B of Vimentin in the Filamentous Form Provides a Model of a High-order Assembly of a Vimentin Filament. FEBS J. 2018, 285, 2888–2899. [Google Scholar] [CrossRef] [Green Version]
- Llères, D.; James, J.; Swift, S.; Norman, D.G.; Lamond, A.I. Quantitative Analysis of Chromatin Compaction in Living Cells Using FLIM-FRET. J. Cell Biol. 2009, 187, 481–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Lu, S.; Shi, Y.; Pan, Y.; Limsakul, P.; Chernov, A.V.; Qiu, J.; Chai, X.; Shi, Y.; Wang, P.; et al. Coordinated Histone Modifications and Chromatin Reorganization in a Single Cell Revealed by FRET Biosensors. Proc. Natl. Acad. Sci. USA 2018, 115, E11681–E11690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.; Kim, W.; Kim, S.; Kim, Y.; Song, Y.; Bilousov, O.; Kim, J.; Lee, T.; Cha, B.; Kim, M.; et al. Phosphorylation by NLK Inhibits YAP-14-3-3-Interactions and Induces Its Nuclear Localization. EMBO Rep. 2017, 18, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Shreberk-Shaked, M.; Oren, M. New Insights into YAP/TAZ Nucleo-cytoplasmic Shuttling: New Cancer Therapeutic Opportunities? Mol. Oncol. 2019, 13, 1335–1341. [Google Scholar] [CrossRef] [Green Version]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden Gate Shuffling: A One-Pot DNA Shuffling Method Based on Type IIs Restriction Enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [Green Version]
- Engler, C.; Kandzia, R.; Marillonnet, S. A One Pot, One Step, Precision Cloning Method with High Throughput Capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [Green Version]
- Engler, C.; Marillonnet, S. Generation of Families of Construct Variants Using Golden Gate Shuffling. Methods Mol. Biol. 2011, 729, 167–181. [Google Scholar]
- Forster_Distance_Calculator. Available online: https://pymolwiki.org/index.php/Forster_distance_calculator (accessed on 17 November 2021).
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Hoelzel, C.A.; Zhang, X. Visualizing and Manipulating Biological Processes by Using HaloTag and SNAP-Tag Technologies. ChemBioChem 2020, 21, 1935–1946. [Google Scholar] [CrossRef]
- Perfilov, M.M.; Gavrikov, A.S.; Lukyanov, K.A.; Mishin, A.S. Transient Fluorescence Labeling: Low Affinity—High Benefits. Int. J. Mol. Sci. 2021, 22, 11799. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrikov, A.S.; Bozhanova, N.G.; Baranov, M.S.; Mishin, A.S. Add and Go: FRET Acceptor for Live-Cell Measurements Modulated by Externally Provided Ligand. Int. J. Mol. Sci. 2022, 23, 4396. https://doi.org/10.3390/ijms23084396
Gavrikov AS, Bozhanova NG, Baranov MS, Mishin AS. Add and Go: FRET Acceptor for Live-Cell Measurements Modulated by Externally Provided Ligand. International Journal of Molecular Sciences. 2022; 23(8):4396. https://doi.org/10.3390/ijms23084396
Chicago/Turabian StyleGavrikov, Alexey S., Nina G. Bozhanova, Mikhail S. Baranov, and Alexander S. Mishin. 2022. "Add and Go: FRET Acceptor for Live-Cell Measurements Modulated by Externally Provided Ligand" International Journal of Molecular Sciences 23, no. 8: 4396. https://doi.org/10.3390/ijms23084396
APA StyleGavrikov, A. S., Bozhanova, N. G., Baranov, M. S., & Mishin, A. S. (2022). Add and Go: FRET Acceptor for Live-Cell Measurements Modulated by Externally Provided Ligand. International Journal of Molecular Sciences, 23(8), 4396. https://doi.org/10.3390/ijms23084396