
VI-116, A Novel Potent Inhibitor of VRAC with Minimal Effect on ANO1


Abstract
:1. Introduction
2. Results
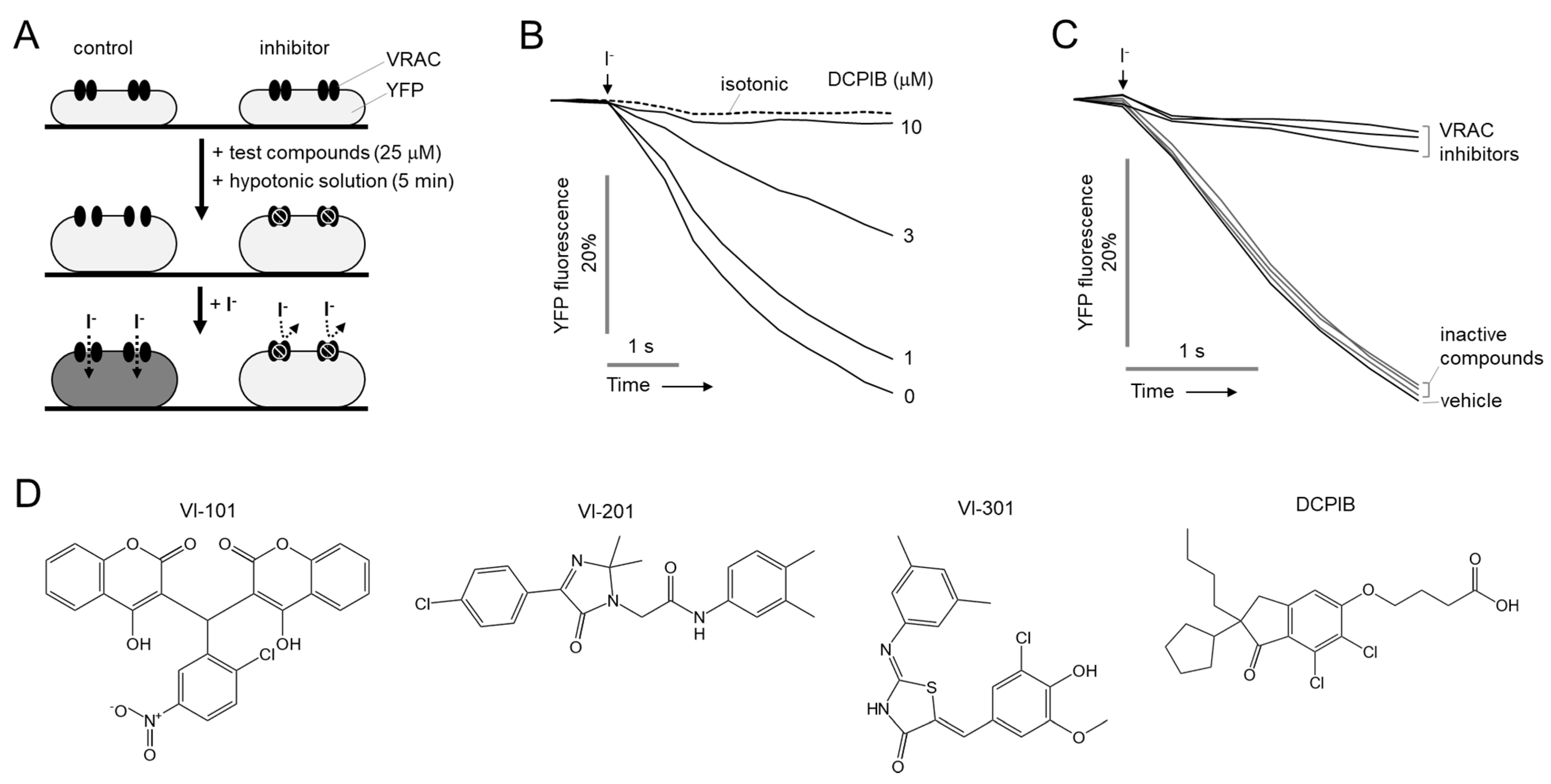
2.1. Identification of Novel Small Molecule Inhibitors of VRAC
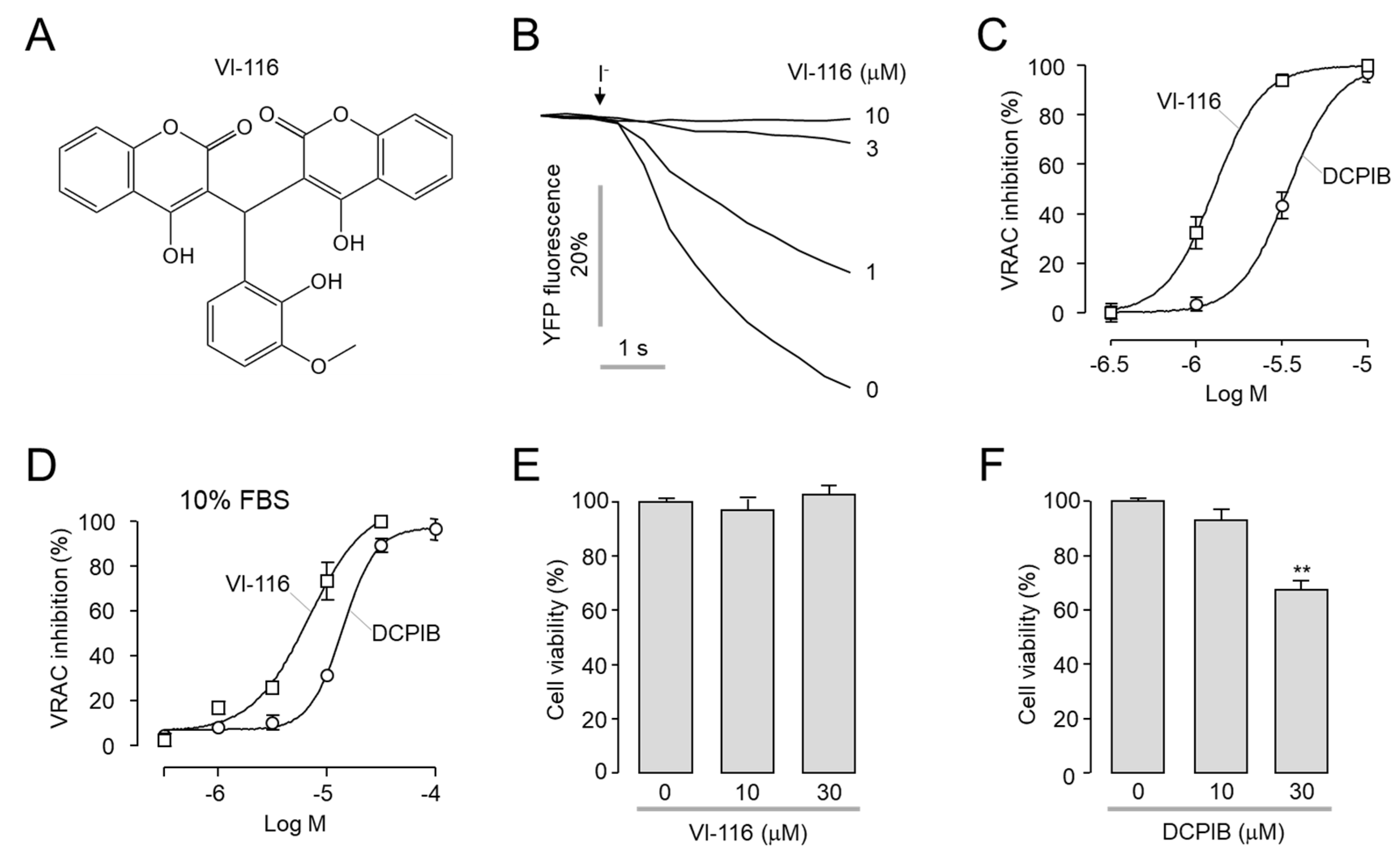
2.2. Identification of a Potent and Selective VRAC Inhibitor, VI-116
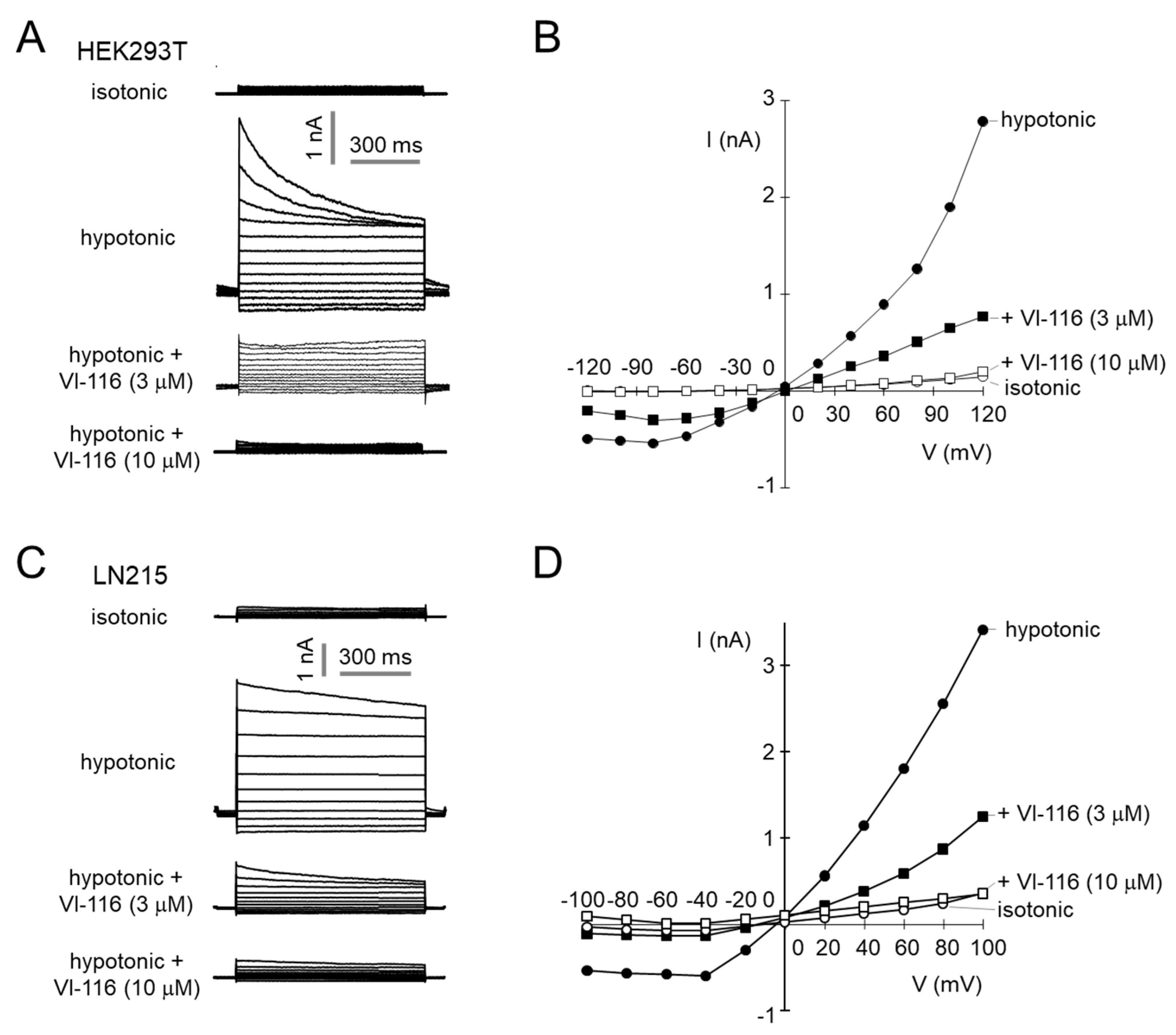
2.3. VI-116 Potently Blocks VRAC-Mediated Chloride Currents
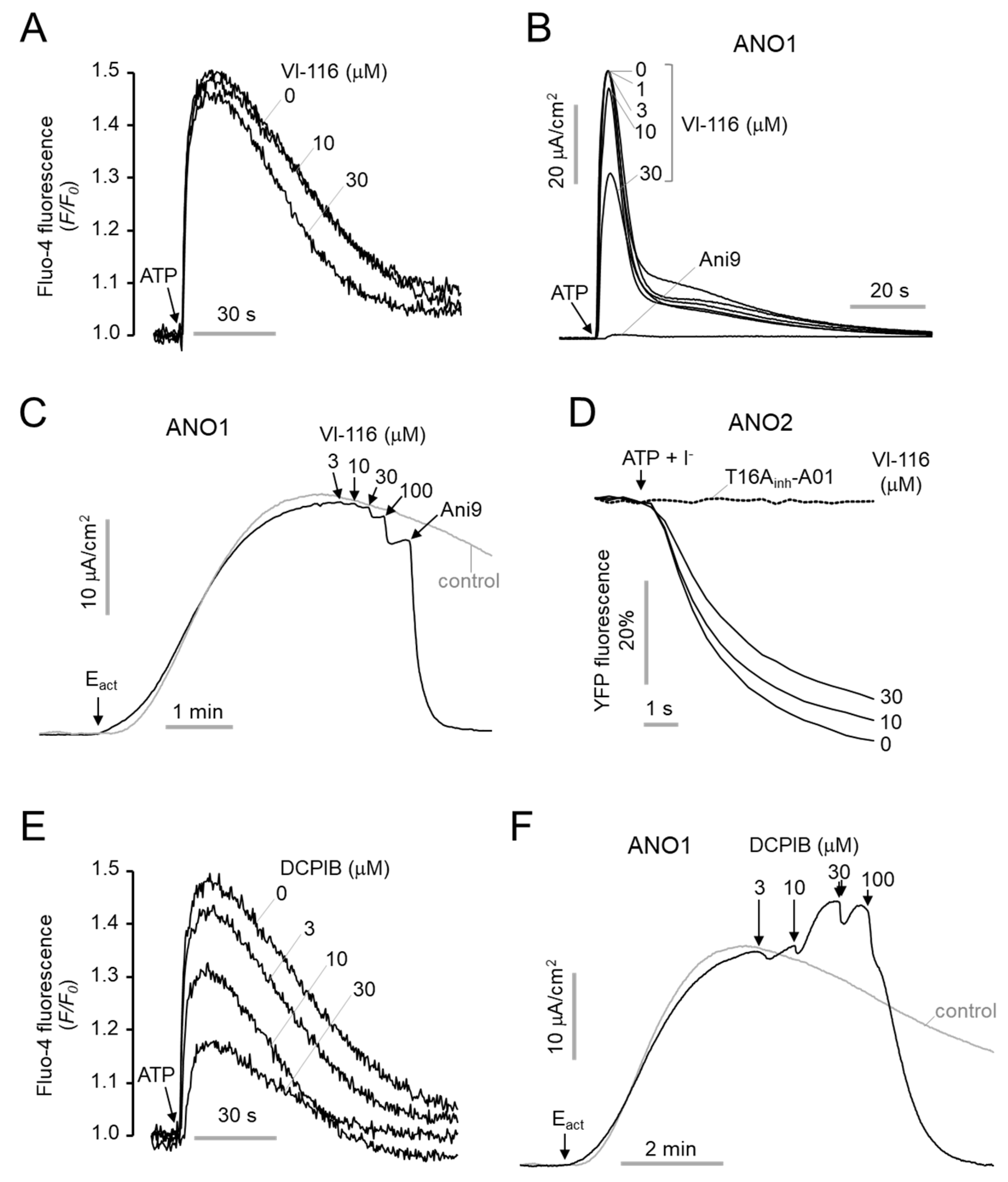
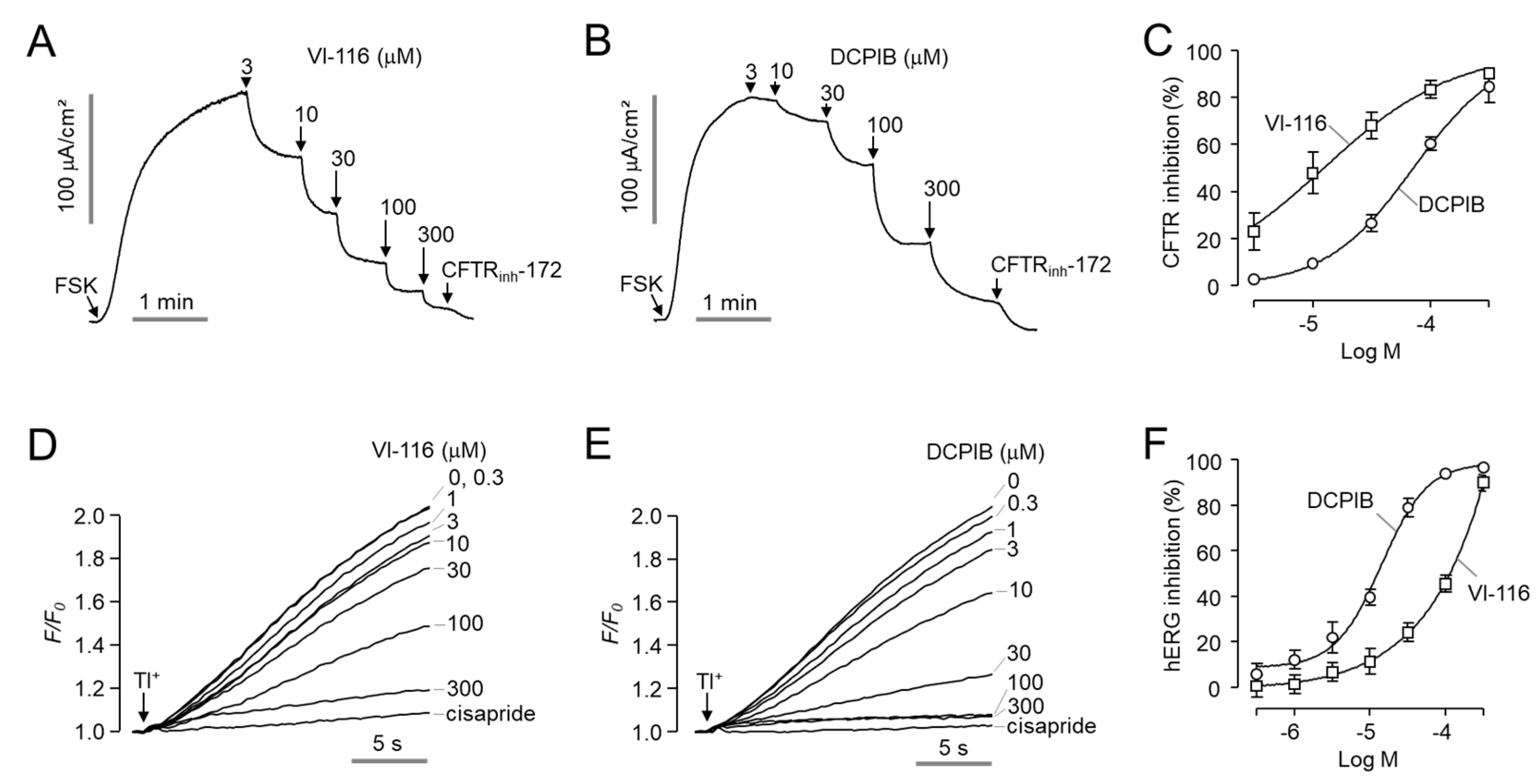
2.4. Minimal Effect of VI-116 on Human ANO1, ANO2 and hERG Channel Activity
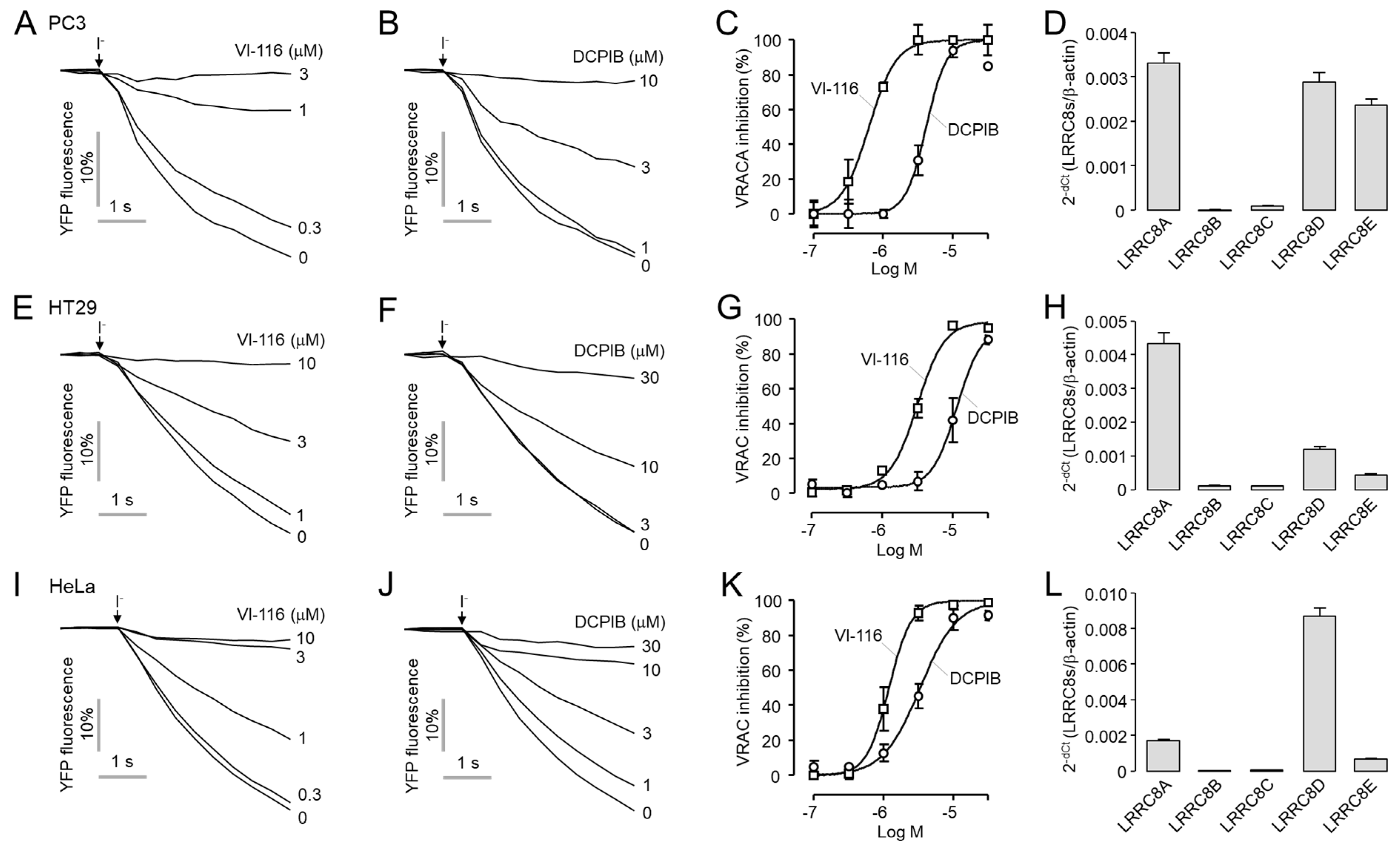
2.5. VI-116 Potently Inhibits Endogenous VRAC Activity in PC3, HT29 and HeLa Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Lines
4.2. Materials and Reagents
4.3. YFP Fluorescence Quenching Assay
4.4. Apical Membrane Current Measurement
4.5. FluxOR Potassium Ion Channel Assay
4.6. Patch-Clamp
4.7. Cell Proliferation Assay
4.8. Quantitative PCR Analysis
4.9. Intracellular Calcium Measurement
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANO1 | anoctamin 1 |
| ANO2 | anoctamin 2 |
| AVD | apoptotic volume decrease |
| GPCR | G protein-coupled receptor |
| hERG | human ether-a-go-go related gene |
| HTS | high-throughput screening |
| RVD | regulatory volume decrease |
| TREK1 | twik-related K+ channel 1 |
| TREK2 | twik-related K+ channel 2 |
| VRAC | volume regulated anion channel |
References
- Pedersen, S.F.; Kapus, A.; Hoffmann, E.K. Osmosensory mechanisms in cellular and systemic volume regulation. J. Am. Soc. Nephrol. 2011, 22, 1587–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. Swell1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, F.K.; Ullrich, F.; Munch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of lrrc8 heteromers as an essential component of the volume-regulated anion channel vrac. Science 2014, 344, 634–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimelberg, H.K. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 2005, 50, 389–397. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of vrac channels determines substrate specificity and cellular resistance to pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Choi, H.; Rohrbough, J.C.; Nguyen, H.N.; Dikalova, A.; Lamb, F.S. Oxidant-resistant lrrc8a/c anion channels support superoxide production by nadph oxidase 1. J. Physiol. 2021, 599, 3013–3036. [Google Scholar] [CrossRef]
- Oh, U.; Jung, J. Cellular functions of tmem16/anoctamin. Pflugers. Arch 2016, 468, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xie, L.; Gunasekar, S.K.; Mishra, A.; Zhang, Y.; Pai, S.; Gao, Y.; Kumar, A.; Norris, A.W.; Stephens, S.B.; et al. Swell1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nat. Commun. 2018, 9, 367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Deng, Z.; Zhang, D.; Li, H.; Zhang, L.; Niu, J.; Zuo, W.; Fu, R.; Fan, L.; Ye, J.H.; et al. High expression of leucinerich repeatcontaining 8a is indicative of a worse outcome of colon cancer patients by enhancing cancer cell growth and metastasis. Oncol. Rep. 2018, 40, 1275–1286. [Google Scholar]
- Katoh, M.; Katoh, M. Flj10261 gene, located within the ccnd1-ems1 locus on human chromosome 11q13, encodes the eight-transmembrane protein homologous to c12orf3, c11orf25 and flj34272 gene products. Int. J. Oncol. 2003, 22, 1375–1381. [Google Scholar] [CrossRef]
- Duvvuri, U.; Shiwarski, D.J.; Xiao, D.; Bertrand, C.; Huang, X.; Edinger, R.S.; Rock, J.R.; Harfe, B.D.; Henson, B.J.; Kunzelmann, K.; et al. Tmem16a induces mapk and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012, 72, 3270–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, C.; Martins, J.R.; Rudin, F.; Schneider, S.; Dietsche, T.; Fischer, C.A.; Tornillo, L.; Terracciano, L.M.; Schreiber, R.; Bubendorf, L.; et al. Enhanced expression of ano1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS ONE 2012, 7, e43265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.; Lee, H.K.; Park, J.; Jeon, D.K.; Jo, S.; Jo, M.; Namkung, W. Ani9, a novel potent small-molecule ano1 inhibitor with negligible effect on ano2. PLoS ONE 2016, 11, e0155771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Sehrer, J.; Viana, F.; De Greef, C.; Raeymaekers, L.; Eggermont, J.; Droogmans, G. Volume-activated Cl− currents in different mammalian non-excitable cell types. Pflugers Arch. 1994, 428, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Prenen, J.; Kamouchi, M.; Viana, F.; Voets, T.; Droogmans, G. Inhibition by mibefradil, a novel calcium channel antagonist, of Ca2+- and volume-activated Cl− channels in macrovascular endothelial cells. Br. J. Pharmacol. 1997, 121, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.F.; Prenen, J.; Droogmans, G.; Hoffmann, E.K.; Nilius, B. Separate swelling- and Ca2+-activated anion currents in ehrlich ascites tumor cells. J. Membr. Biol. 1998, 163, 97–110. [Google Scholar] [CrossRef]
- Maertens, C.; Wei, L.; Voets, T.; Droogmans, G.; Nilius, B. Block by fluoxetine of volume-regulated anion channels. Br. J. Pharmacol. 1999, 126, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Maertens, C.; Wei, L.; Droogmans, G.; Nilius, B. Inhibition of volume-regulated and calcium-activated chloride channels by the antimalarial mefloquine. J. Pharmacol. Exp. Ther. 2000, 295, 29–36. [Google Scholar]
- Maertens, C.; Droogmans, G.; Chakraborty, P.; Nilius, B. Inhibition of volume-regulated anion channels in cultured endothelial cells by the anti-oestrogens clomiphene and nafoxidine. Br. J. Pharmacol. 2001, 132, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, V.; Caprini, M.; Nicchia, G.P.; Rossi, A.; Dovizio, M.; Cervetto, C.; Nobile, M.; Ferroni, S. Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels 2009, 3, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, E.E.; Kramer, M.; Strange, K.; Denton, J.S. Cyslt1 receptor antagonists pranlukast and zafirlukast inhibit lrrc8-mediated volume regulated anion channels independently of the receptor. Am. J. Physiol. Cell Physiol. 2019, 317, C857–C866. [Google Scholar] [CrossRef] [PubMed]
- Bowens, N.H.; Dohare, P.; Kuo, Y.H.; Mongin, A.A. Dcpib, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol. Pharmacol. 2013, 83, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minieri, L.; Pivonkova, H.; Caprini, M.; Harantova, L.; Anderova, M.; Ferroni, S. The inhibitor of volume-regulated anion channels dcpib activates trek potassium channels in cultured astrocytes. Br. J. Pharmacol. 2013, 168, 1240–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolini, P.; Ferrera, L.; Remigante, A.; Picco, C.; Barbieri, R.; Bertelli, S.; Moran, O.; Gavazzo, P.; Pusch, M. The vrac blocker dcpib directly gates the bk channels and increases intracellular Ca2+ in melanoma and pancreatic duct adenocarcinoma cell lines. Br. J. Pharmacol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Namkung, W.; Yao, Z.; Finkbeiner, W.E.; Verkman, A.S. Small-molecule activators of tmem16a, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011, 25, 4048–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.D.; Singleton, C.B.; Bursill, J.A.; Wyse, K.R.; Valenzuela, S.M.; Qiu, M.R.; Breit, S.N.; Campbell, T.J. Inhibition of the human ether-a-go-go-related gene (herg) potassium channel by cisapride: Affinity for open and inactivated states. Br. J. Pharmacol. 1999, 128, 444–450. [Google Scholar] [CrossRef]
- Jentsch, T.J. Vracs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef]
- Benedetto, R.; Sirianant, L.; Pankonien, I.; Wanitchakool, P.; Ousingsawat, J.; Cabrita, I.; Schreiber, R.; Amaral, M.; Kunzelmann, K. Relationship between tmem16a/anoctamin 1 and lrrc8a. Pflugers Arch. 2016, 468, 1751–1763. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Men, H.; Zhang, F.; Zhang, H. Lrrca8a and ano1 contribute to serum-induced vrac in a Ca2+-dependent manners. J. Pharmacol. Sci. 2020, 143, 176–181. [Google Scholar] [CrossRef]
- Decher, N.; Lang, H.J.; Nilius, B.; Bruggemann, A.; Busch, A.E.; Steinmeyer, K. Dcpib is a novel selective blocker of i(cl, swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br. J. Pharmacol. 2001, 134, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Friard, J.; Tauc, M.; Cougnon, M.; Compan, V.; Duranton, C.; Rubera, I. Comparative effects of chloride channel inhibitors on lrrc8/vrac-mediated chloride conductance. Front. Pharmacol. 2017, 8, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Matsuda, M.; Kato, K.; Jimi, E.; Takeuchi, H.; Nakano, S.; Kajioka, S.; Matsuzaki, E.; Hirofuji, T.; Inoue, R.; et al. Volume-regulated chloride channel regulates cell proliferation and is involved in the possible interaction between tmem16a and lrrc8a in human metastatic oral squamous cell carcinoma cells. Eur. J. Pharmacol. 2021, 895, 173881. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
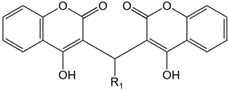 | |||||||
|---|---|---|---|---|---|---|---|
| ID | R1 | VRAC IC50 (μM) | ANO1 IC50 (μM) | ID | R1 | VRAC IC50 (μM) | ANO1 IC50 (μM) |
| VI-101 | 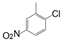 | 3.3 | 3.2 | VI-111 |  | 2.9 | 3.2 |
| VI-102 | 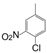 | 0.8 | 3.0 | VI-112 | 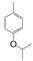 | 1.4 | 0.8 |
| VI-103 |  | 3.3 | 3.2 | VI-113 | 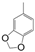 | 1.9 | 3.9 |
| VI-104 | 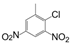 | 30.0 | 37.9 | VI-114 | 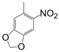 | 10.6 | 20.3 |
| VI-105 |  | 4.9 | 3.8 | VI-115 | 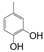 | 47.9 | 61.6 |
| VI-106 | 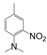 | 1.3 | 3.6 | VI-116 | 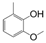 | 1.3 | 39.1 |
| VI-107 | 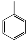 | 103.3 | 95.2 | VI-117 |  | 6.8 | 14.2 |
| VI-108 | 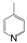 | 17.0 | 34.7 | VI-118 | 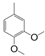 | 4.0 | 14.2 |
| VI-109 | 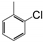 | 0.9 | 2.0 | VI-119 | 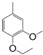 | 12.0 | 4.9 |
| VI-110 | 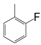 | 0.6 | 4.8 | VI-120 | 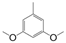 | 9.3 | 3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, D.; Ryu, K.; Jo, S.; Kim, I.; Namkung, W. VI-116, A Novel Potent Inhibitor of VRAC with Minimal Effect on ANO1. Int. J. Mol. Sci. 2022, 23, 5168. https://doi.org/10.3390/ijms23095168
Jeon D, Ryu K, Jo S, Kim I, Namkung W. VI-116, A Novel Potent Inhibitor of VRAC with Minimal Effect on ANO1. International Journal of Molecular Sciences. 2022; 23(9):5168. https://doi.org/10.3390/ijms23095168
Chicago/Turabian StyleJeon, Dongkyu, Kunhi Ryu, Sungwoo Jo, Ikyon Kim, and Wan Namkung. 2022. "VI-116, A Novel Potent Inhibitor of VRAC with Minimal Effect on ANO1" International Journal of Molecular Sciences 23, no. 9: 5168. https://doi.org/10.3390/ijms23095168
APA StyleJeon, D., Ryu, K., Jo, S., Kim, I., & Namkung, W. (2022). VI-116, A Novel Potent Inhibitor of VRAC with Minimal Effect on ANO1. International Journal of Molecular Sciences, 23(9), 5168. https://doi.org/10.3390/ijms23095168