
Phosphoproteomics Unravel HBV Triggered Rewiring of Host Phosphosignaling Events

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Altered Phosphoproteome Landscape during HBV-Infection
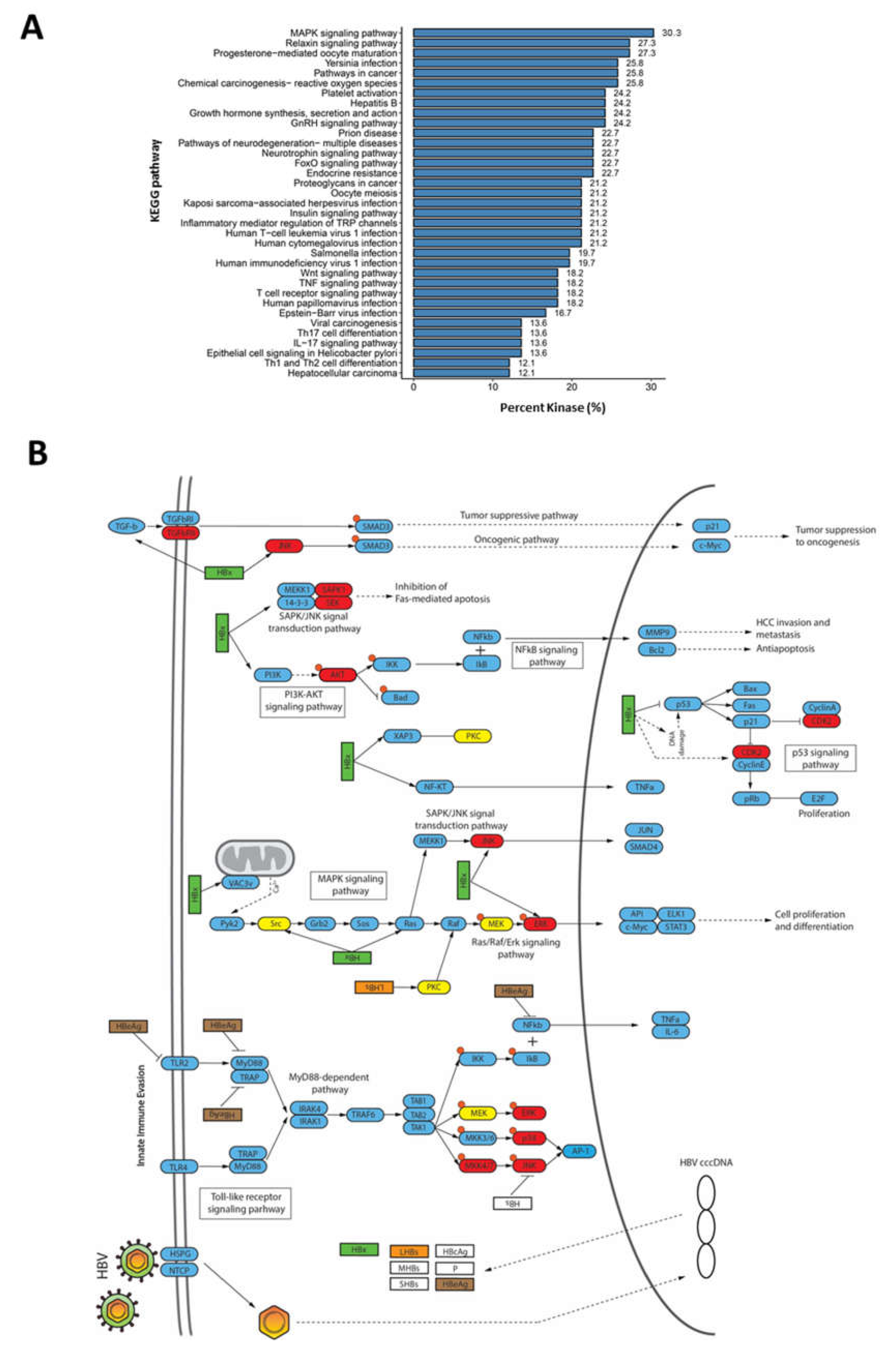
2.2. Dysregulation of Biological Processes during HBV Infection
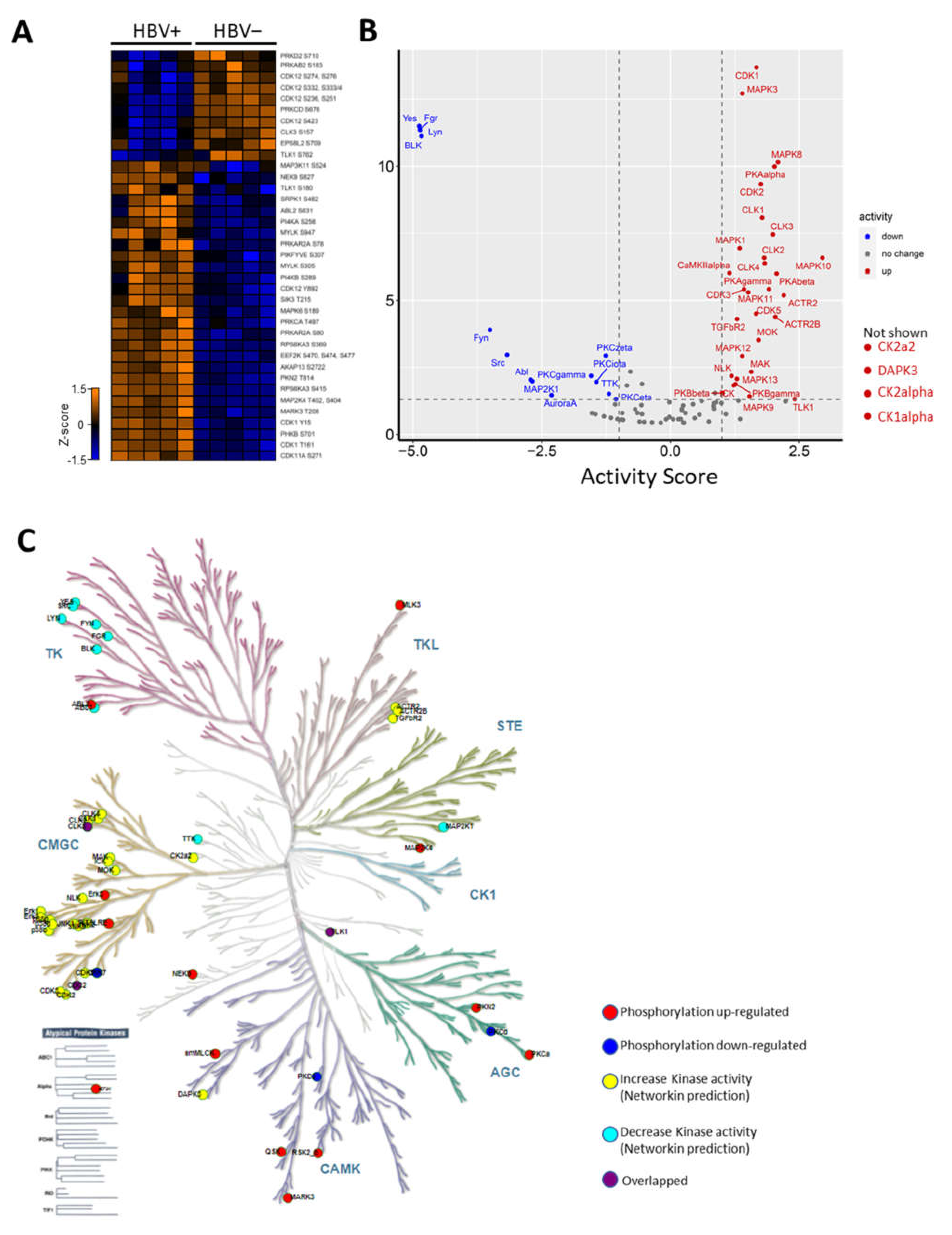
2.3. HBV Infection-Induced Host Kinome
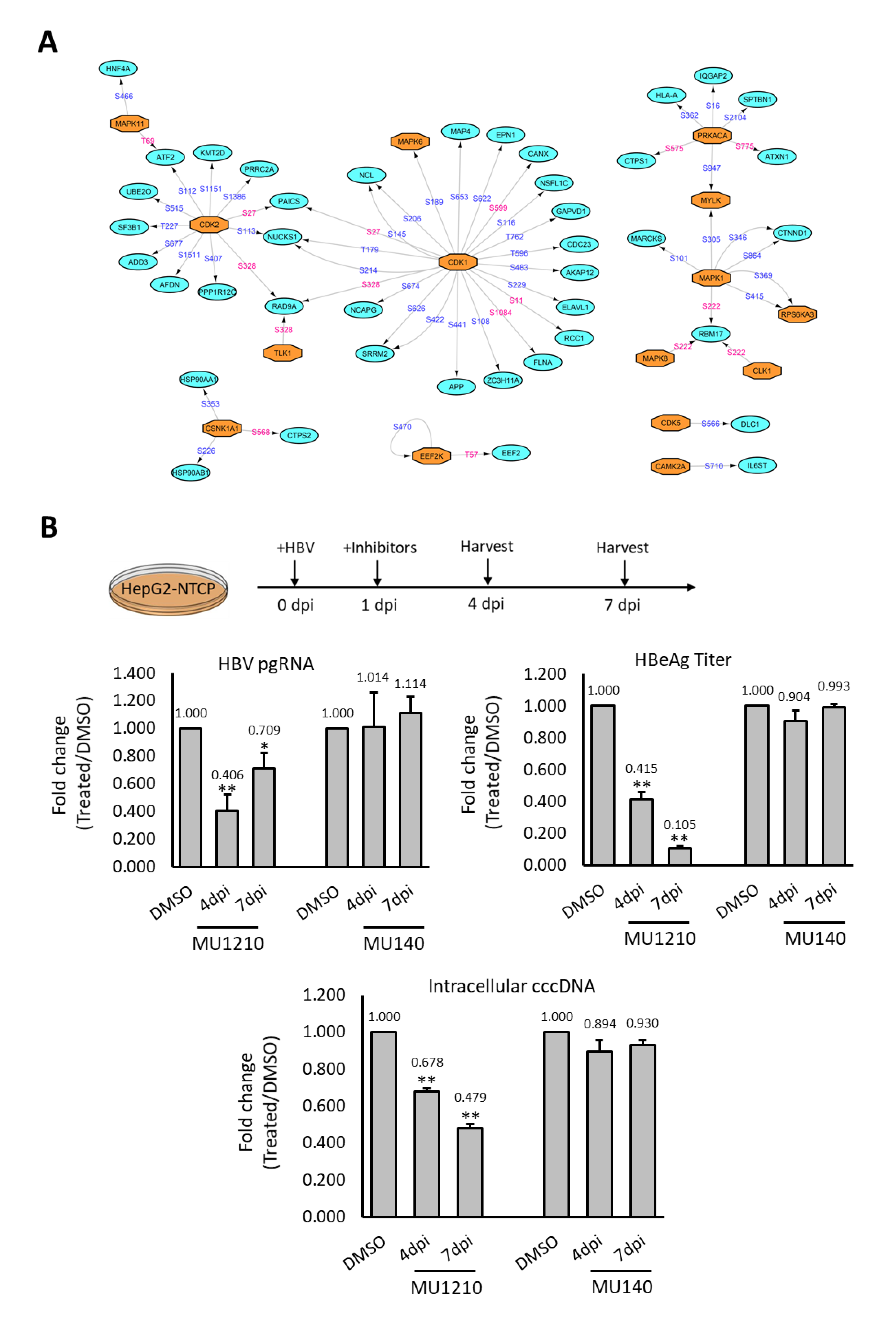
2.4. Rewiring of Host Phosphosignaling Pathway during HBV Infection
2.5. Assessing Deregulated Kinases as Therapeutic Targets for HBV
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. HBV Infection
4.3. Extraction of RNA and DNA
4.4. Real-Time Quantitative PCR and HBeAg ELISA
4.5. Immunofluorescence
4.6. Cell Lysate Preparation
4.7. Reduction, Alkylation and Digestion
4.8. Tandem Mass Tag (TMT) Labelling
4.9. Phosphopeptide Enrichment
4.10. Mass Spectrometry Data Acquisition
4.11. Identification and Quantification of Phosphopeptides
4.12. Analysis of Differentially Regulated Phosphoproteins
4.13. Kinase–Substrate Prediction and Enrichment Assay
4.14. Cytotoxicity Assay
4.15. Kinase Inhibitor Treatment of HBV Infected HepG2-NTCP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed]
- The Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Yang, F. Post-translational Modification Control of HBV Biological Processes. Front. Microbiol. 2018, 9, 2661. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Sawano, Y.; Kosuge, S.; Yamano, Y.; Kuroki, K.; Ohtsuki, K. High phosphorylation of HBV core protein by two alpha-type CK2-activated cAMP-dependent protein kinases in vitro. FEBS Lett. 2006, 580, 894–899. [Google Scholar] [CrossRef]
- Daub, H.; Blencke, S.; Habenberger, P.; Kurtenbach, A.; Dennenmoser, J.; Wissing, J.; Ullrich, A.; Cotten, M. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J. Virol. 2002, 76, 8124–8137. [Google Scholar] [CrossRef]
- Heger-Stevic, J.; Zimmermann, P.; Lecoq, L.; Bottcher, B.; Nassal, M. Hepatitis B virus core protein phosphorylation: Identification of the SRPK1 target sites and impact of their occupancy on RNA binding and capsid structure. PLoS Pathog. 2018, 14, e1007488. [Google Scholar] [CrossRef]
- Liu, K.; Ludgate, L.; Yuan, Z.; Hu, J. Regulation of multiple stages of hepadnavirus replication by the carboxyl-terminal domain of viral core protein in trans. J. Virol. 2015, 89, 2918–2930. [Google Scholar] [CrossRef]
- Luo, X.; Zhang, R.; Lu, M.; Liu, S.; Baba, H.A.; Gerken, G.; Wedemeyer, H.; Broering, R. Hippo Pathway Counter-Regulates Innate Immunity in Hepatitis B Virus Infection. Front. Immunol. 2021, 12, 684424. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Suhail, M.; Abdel-Hafiz, H.; Ali, A.; Fatima, K.; Damanhouri, G.A.; Azhar, E.; Chaudhary, A.G.; Qadri, I. Potential mechanisms of hepatitis B virus induced liver injury. World J. Gastroenterol. 2014, 20, 12462–12472. [Google Scholar] [CrossRef] [PubMed]
- Wiredja, D.D.; Koyuturk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef]
- Ren, L.; Li, C.; Wang, Y.; Teng, Y.; Sun, H.; Xing, B.; Yang, X.; Jiang, Y.; He, F. In Vivo Phosphoproteome Analysis Reveals Kinome Reprogramming in Hepatocellular Carcinoma. Mol. Cell. Proteom. 2018, 17, 1067–1083. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Selzer, L.; Kant, R.; Wang, J.C.; Bothner, B.; Zlotnick, A. Hepatitis B Virus Core Protein Phosphorylation Sites Affect Capsid Stability and Transient Exposure of the C-terminal Domain. J. Biol. Chem. 2015, 290, 28584–28593. [Google Scholar] [CrossRef]
- Eid, S.; Turk, S.; Volkamer, A.; Rippmann, F.; Fulle, S. KinMap: A web-based tool for interactive navigation through human kinome data. BMC Bioinform. 2017, 18, 16. [Google Scholar] [CrossRef]
- Megahed, F.A.K.; Zhou, X.; Sun, P. The Interactions between HBV and the Innate Immunity of Hepatocytes. Viruses 2020, 12, 285. [Google Scholar] [CrossRef]
- Kim, S.; Lee, H.S.; Ji, J.H.; Cho, M.Y.; Yoo, Y.S.; Park, Y.Y.; Cha, H.J.; Lee, Y.; Kim, Y.; Cho, H. Hepatitis B virus X protein activates the ATM-Chk2 pathway and delays cell cycle progression. J. Gen. Virol. 2015, 96, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Panteva, M.; Korkaya, H.; Jameel, S. Hepatitis viruses and the MAPK pathway: Is this a survival strategy? Virus Res. 2003, 92, 131–140. [Google Scholar] [CrossRef]
- Nemec, V.; Maier, L.; Berger, B.T.; Chaikuad, A.; Drapela, S.; Soucek, K.; Knapp, S.; Paruch, K. Highly selective inhibitors of protein kinases CLK and HIPK with the furo [3,2-b]pyridine core. Eur. J. Med. Chem. 2021, 215, 113299. [Google Scholar] [CrossRef] [PubMed]
- Martin Moyano, P.; Nemec, V.; Paruch, K. Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. Int. J. Mol. Sci. 2020, 21, 7549. [Google Scholar] [CrossRef]
- Lupberger, J.; Schaedler, S.; Peiran, A.; Hildt, E. Identification and characterization of a novel bipartite nuclear localization signal in the hepatitis B virus polymerase. World J. Gastroenterol. 2013, 19, 8000–8010. [Google Scholar] [CrossRef]
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef]
- Pastor, F.; Shkreta, L.; Chabot, B.; Durantel, D.; Salvetti, A. Interplay Between CMGC Kinases Targeting SR Proteins and Viral Replication: Splicing and Beyond. Front. Microbiol. 2021, 12, 658721. [Google Scholar] [CrossRef]
- Mylonis, I.; Giannakouros, T. Protein kinase CK2 phosphorylates and activates the SR protein-specific kinase 1. Biochem. Biophys. Res. Commun. 2003, 301, 650–656. [Google Scholar] [CrossRef]
- Li, Y.X.; Ren, Y.L.; Fu, H.J.; Zou, L.; Yang, Y.; Chen, Z. Hepatitis B Virus Middle Protein Enhances IL-6 Production via p38 MAPK/NF-κB Pathways in an ER Stress-Dependent Manner. PLoS ONE 2016, 11, e0159089. [Google Scholar] [CrossRef]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar] [CrossRef]
- Cheng, P.; Li, Y.; Yang, L.; Wen, Y.; Shi, W.; Mao, Y.; Chen, P.; Lv, H.; Tang, Q.; Wei, Y. Hepatitis B virus X protein (HBx) induces G2/M arrest and apoptosis through sustained activation of cyclin B1-CDK1 kinase. Oncol. Rep. 2009, 22, 1101–1107. [Google Scholar] [PubMed]
- Xia, Y.; Cheng, X.; Li, Y.; Valdez, K.; Chen, W.; Liang, T.J. Hepatitis B Virus Deregulates the Cell Cycle To Promote Viral Replication and a Premalignant Phenotype. J. Virol. 2018, 92, e00722-18. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef]
- Jiang, Z.; Ninomiya-Tsuji, J.; Qian, Y.; Matsumoto, K.; Li, X. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol. Cell. Biol. 2002, 22, 7158–7167. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Song, H.; Tan, G.; Yang, Y.; Cui, A.; Li, H.; Li, T.; Wu, Z.; Yang, M.; Lv, G.; Chi, X.; et al. Hepatitis B Virus-Induced Imbalance of Inflammatory and Antiviral Signaling by Differential Phosphorylation of STAT1 in Human Monocytes. J. Immunol. 2019, 202, 2266–2275. [Google Scholar] [CrossRef]
- Oh, H.; Irvine, K.D. In vivo regulation of Yorkie phosphorylation and localization. Development 2008, 135, 1081–1088. [Google Scholar] [CrossRef]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Moeslein, F.M.; Myers, M.P.; Landreth, G.E. The CLK family kinases, CLK1 and CLK2, phosphorylate and activate the tyrosine phosphatase, PTP-1B. J. Biol. Chem. 1999, 274, 26697–26704. [Google Scholar] [CrossRef] [PubMed]
- Colwill, K.; Feng, L.L.; Yeakley, J.M.; Gish, G.D.; Cáceres, J.F.; Pawson, T.; Fu, X.D. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J. Biol. Chem. 1996, 271, 24569–24575. [Google Scholar] [CrossRef] [PubMed]
- Uzor, S.; Porazinski, S.R.; Li, L.; Clark, B.; Ajiro, M.; Iida, K.; Hagiwara, M.; Alqasem, A.A.; Perks, C.M.; Wilson, I.D.; et al. CDC2-like (CLK) protein kinase inhibition as a novel targeted therapeutic strategy in prostate cancer. Sci. Rep. 2021, 11, 7963. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Balachandran, A.; Mao, A.Y.; Dobson, W.; Gray-Owen, S.; Cochrane, A. Differential effect of CLK SR Kinases on HIV-1 gene expression: Potential novel targets for therapy. Retrovirology 2011, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Kremsdorf, D.; Lekbaby, B.; Bablon, P.; Sotty, J.; Augustin, J.; Schnuriger, A.; Pol, J.; Soussan, P. Alternative splicing of viral transcripts: The dark side of HBV. Gut 2021, 70, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014, 443, 808–813. [Google Scholar] [CrossRef]
- Ogura, N.; Watashi, K.; Noguchi, T.; Wakita, T. Formation of covalently closed circular DNA in Hep38.7-Tet cells, a tetracycline inducible hepatitis B virus expression cell line. Biochem. Biophys. Res. Commun. 2014, 452, 315–321. [Google Scholar] [CrossRef]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef]
- Jiang, X.; Bomgarden, R.; Brown, J.; Drew, D.L.; Robitaille, A.M.; Viner, R.; Huhmer, A.R. Sensitive and Accurate Quantitation of Phosphopeptides Using TMT Isobaric Labeling Technique. J. Proteome Res. 2017, 16, 4244–4252. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: An integrated platform for kinome biology studies. Nat. Methods 2014, 11, 603–604. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinases Inhibitor | Target | Reported Ki | IC50 |
|---|---|---|---|
| DMAT | CK2 | 40 nM | >500 µM |
| RO-3306 | CDK1 | 35 nM | 164 µM |
| PNU112455A11 | CDK2 and CDK5 | 3.2/3.6 µM (CDK2/5) | >500 µM |
| MU1210 | CLKs | 23 nM | 16.7 mM |
| MU140 | NA (Structural analog of NU1210) | NA | >5 00 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Z.; Mohd-Ismail, N.K.B.; Png, E.; Sze, C.W.; Lin, Q.; Hong, W.; Lim, S.G.; Tan, Y.-J.; Gunaratne, J. Phosphoproteomics Unravel HBV Triggered Rewiring of Host Phosphosignaling Events. Int. J. Mol. Sci. 2022, 23, 5127. https://doi.org/10.3390/ijms23095127
Lim Z, Mohd-Ismail NKB, Png E, Sze CW, Lin Q, Hong W, Lim SG, Tan Y-J, Gunaratne J. Phosphoproteomics Unravel HBV Triggered Rewiring of Host Phosphosignaling Events. International Journal of Molecular Sciences. 2022; 23(9):5127. https://doi.org/10.3390/ijms23095127
Chicago/Turabian StyleLim, ZiJie, Nur Khairiah Binte Mohd-Ismail, Evelyn Png, Ching Wooen Sze, Qifeng Lin, Wanjin Hong, Seng Gee Lim, Yee-Joo Tan, and Jayantha Gunaratne. 2022. "Phosphoproteomics Unravel HBV Triggered Rewiring of Host Phosphosignaling Events" International Journal of Molecular Sciences 23, no. 9: 5127. https://doi.org/10.3390/ijms23095127
APA StyleLim, Z., Mohd-Ismail, N. K. B., Png, E., Sze, C. W., Lin, Q., Hong, W., Lim, S. G., Tan, Y.-J., & Gunaratne, J. (2022). Phosphoproteomics Unravel HBV Triggered Rewiring of Host Phosphosignaling Events. International Journal of Molecular Sciences, 23(9), 5127. https://doi.org/10.3390/ijms23095127