
Lost by Transcription: Fork Failures, Elevated Expression, and Clinical Consequences Related to Deletions in Metastatic Colorectal Cancer


Abstract
:1. Introduction
2. Results
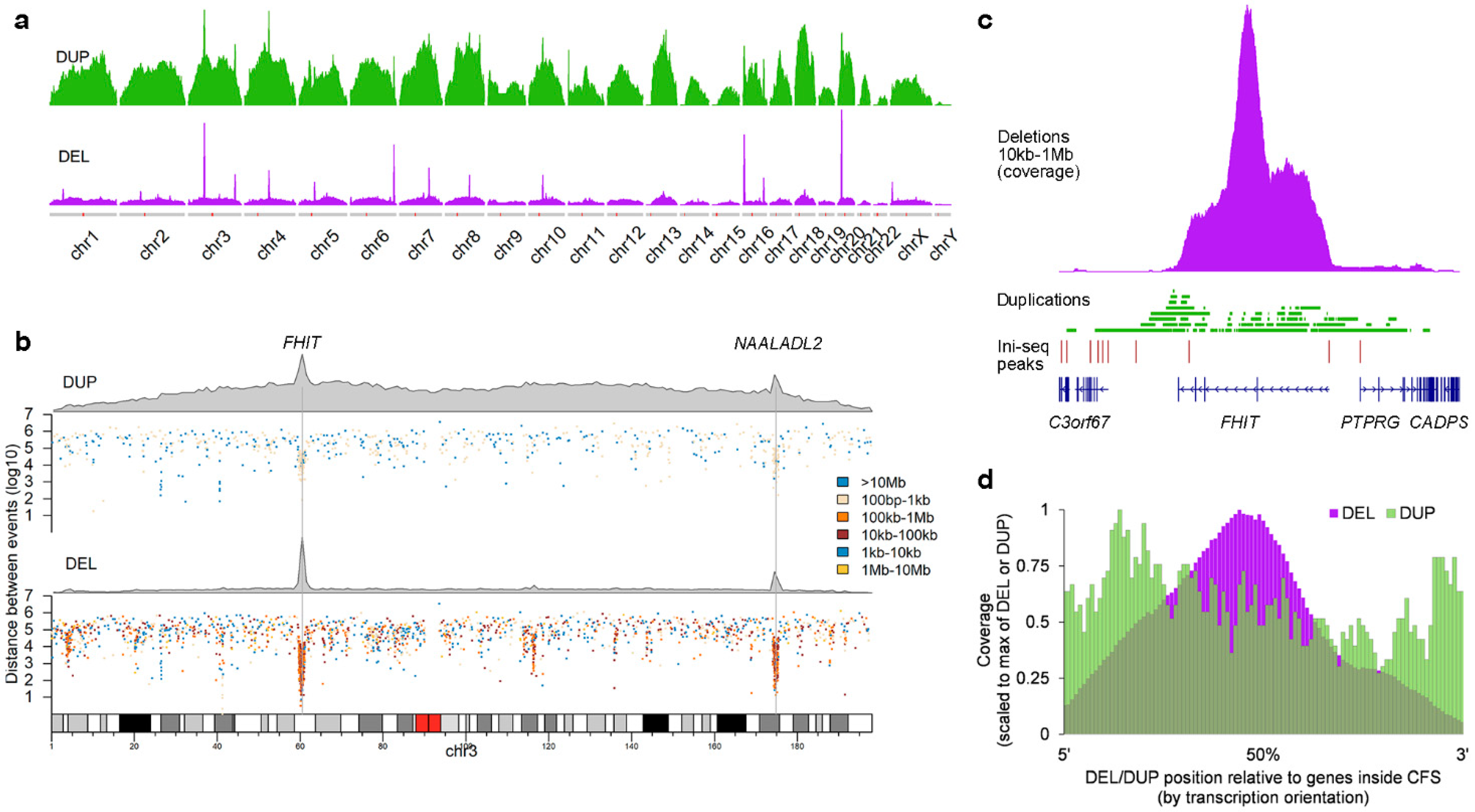
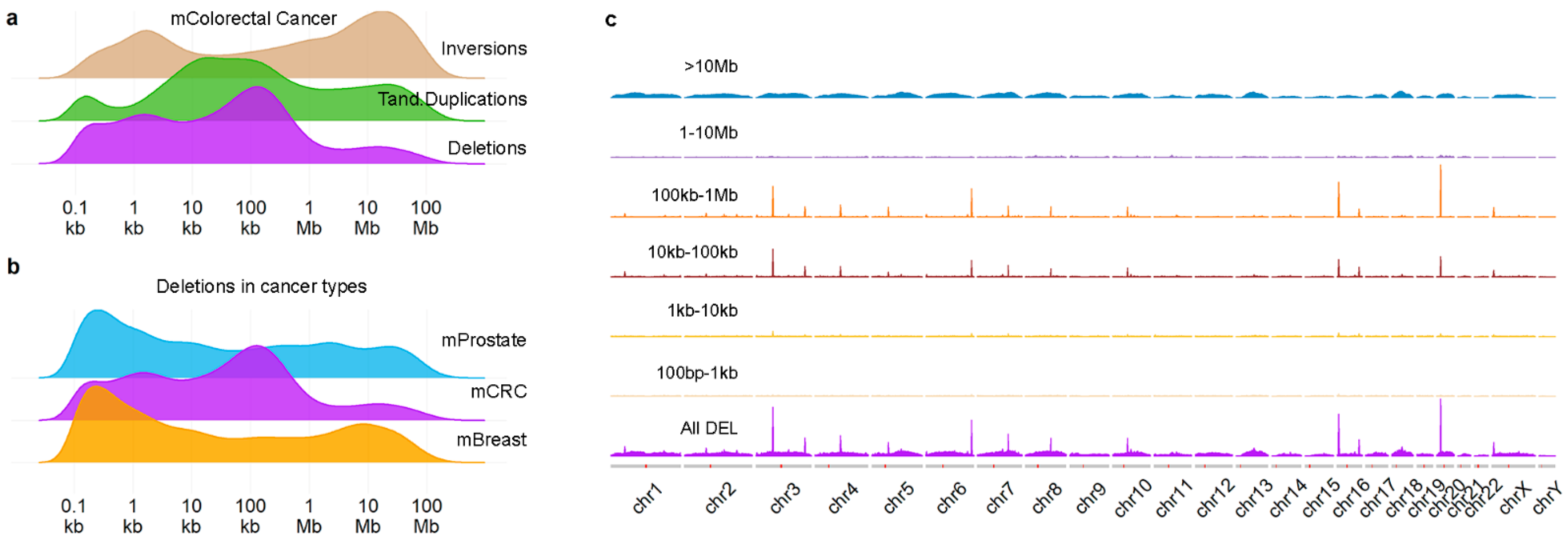
2.1. Position and Size of Chromosomal Deletions in Metastatic mCRC
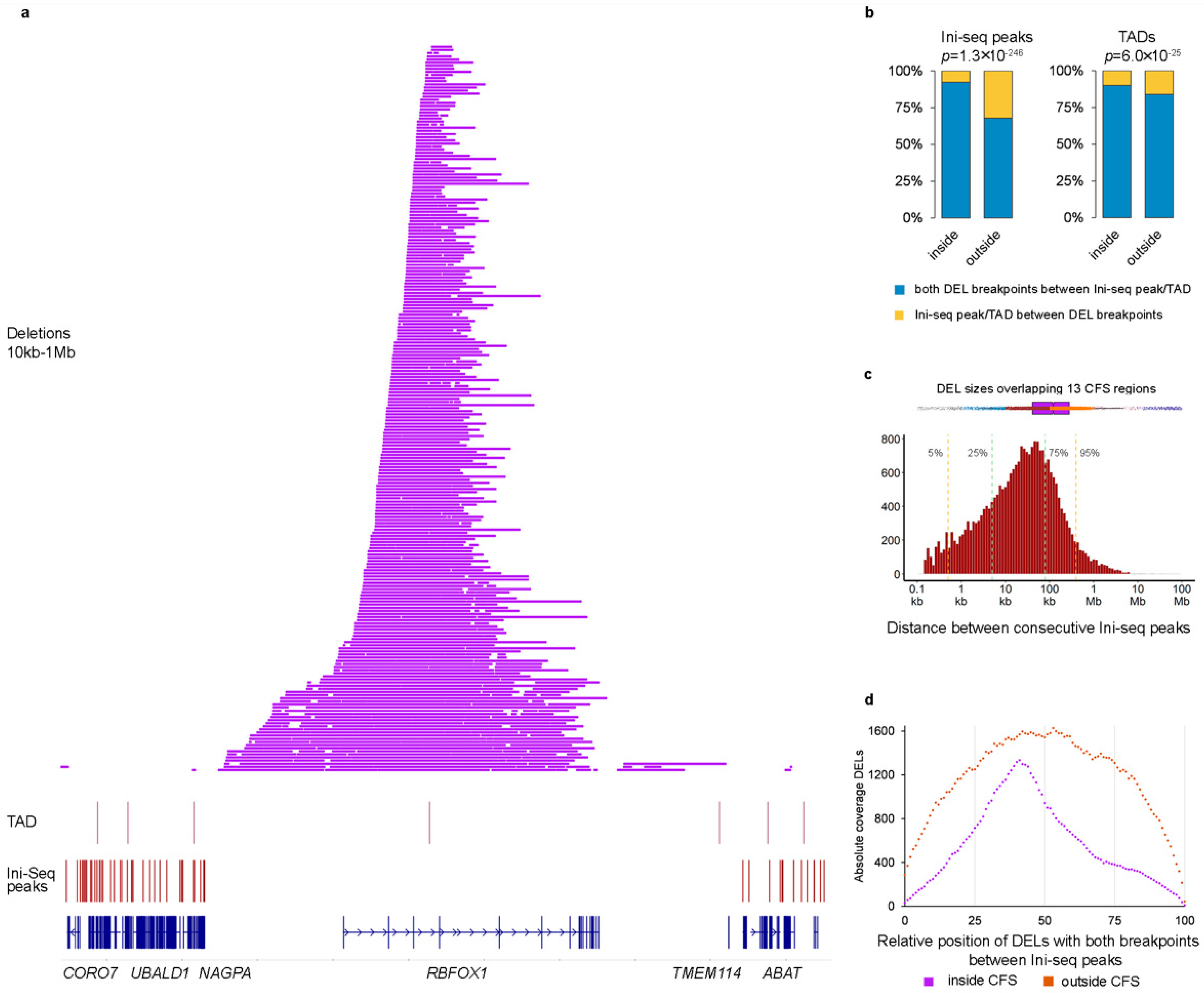
2.2. Deletion and Duplication Patterns Concur with the Transcription-Dependent Double-Fork Failure Model
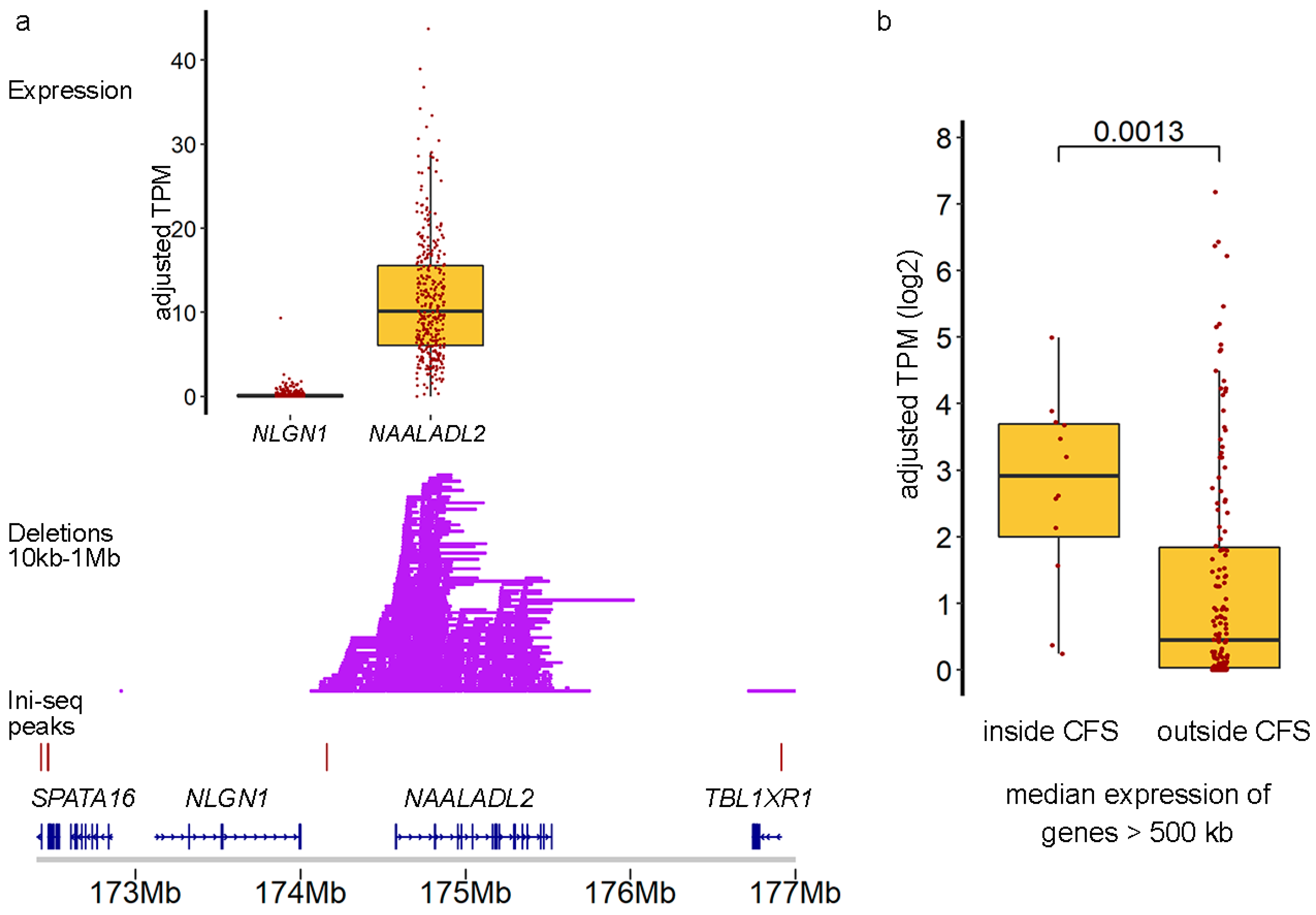
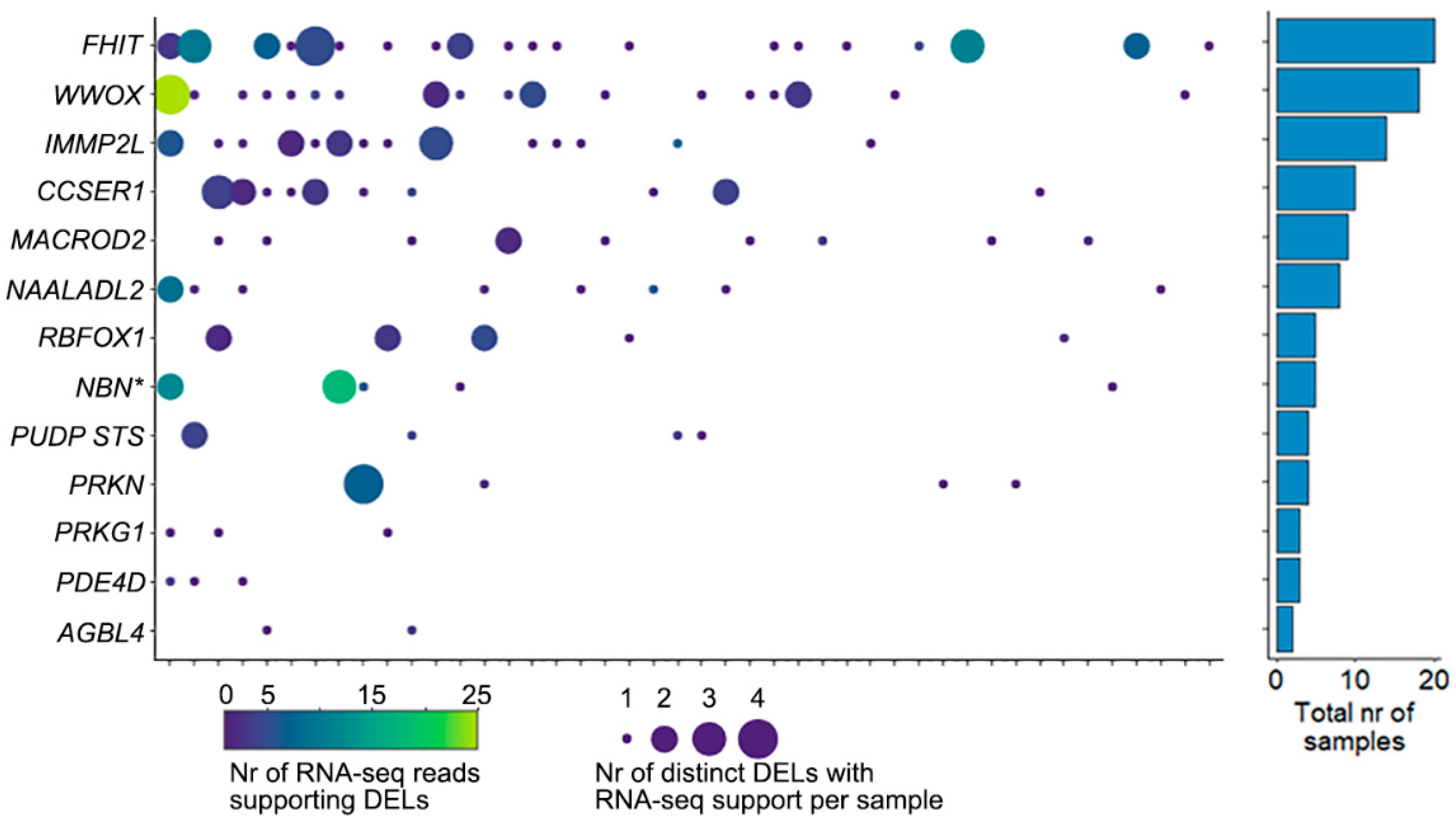
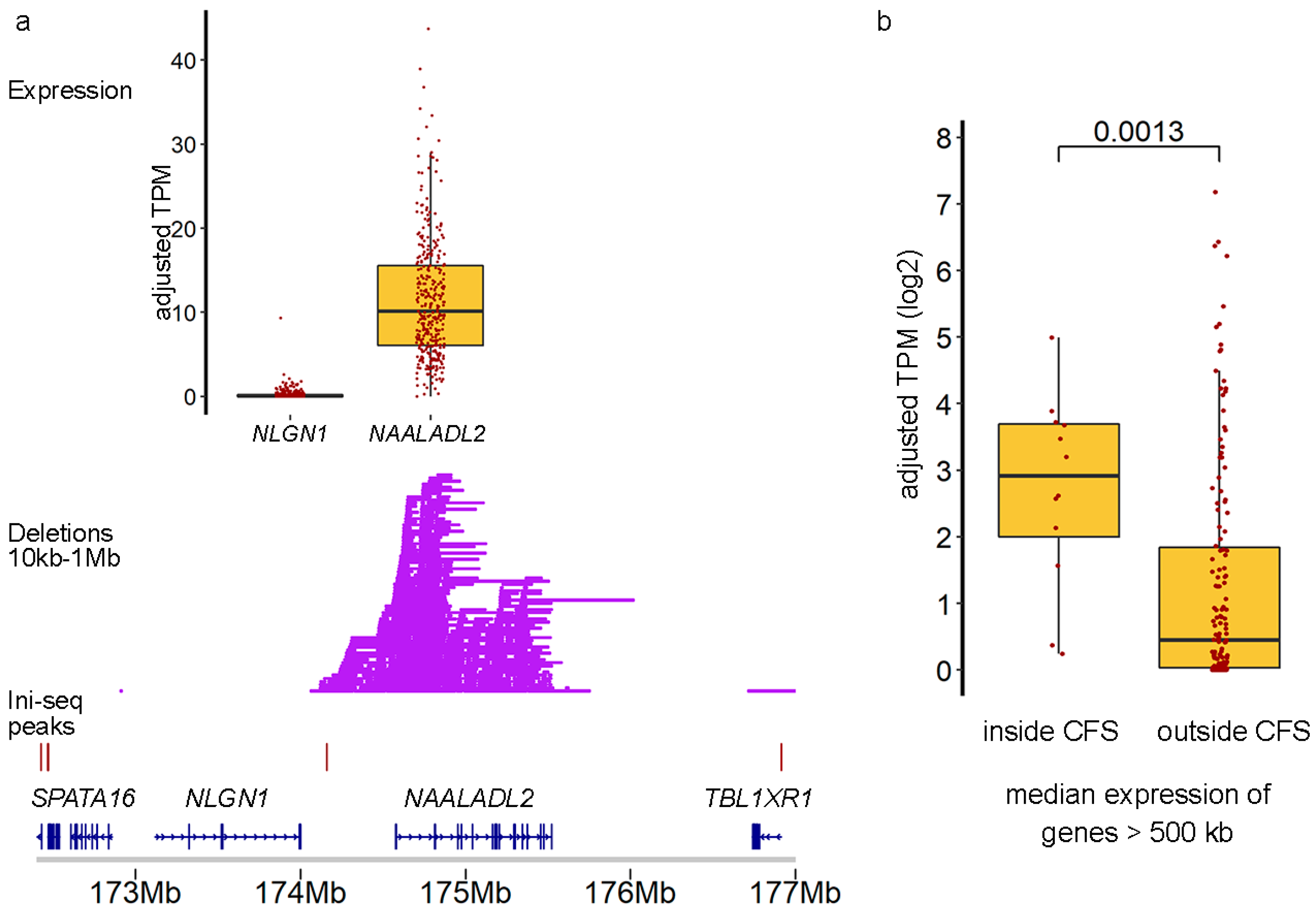
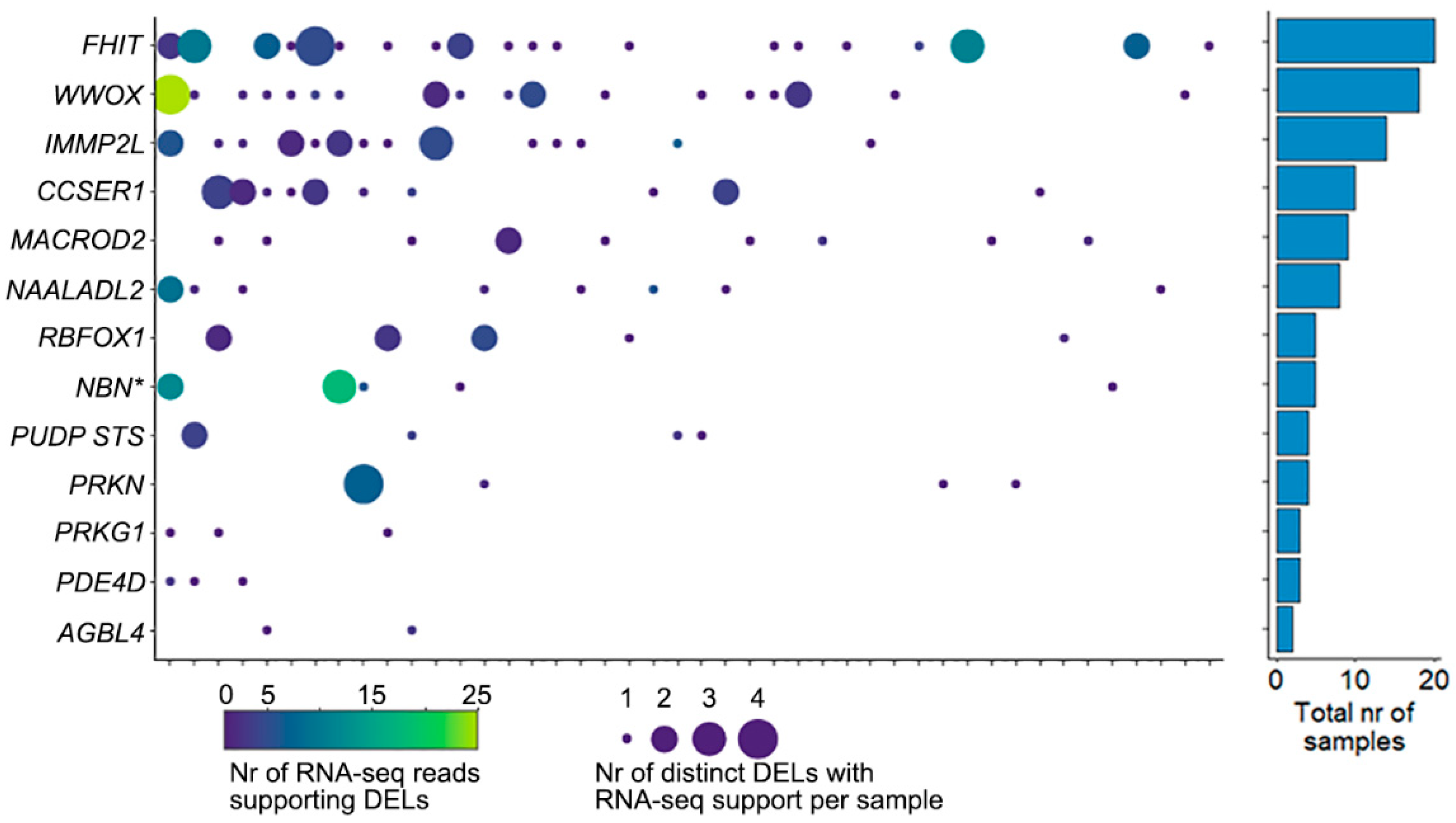
2.3. Cells with DELs in CFS Retain Transcription of the Locus
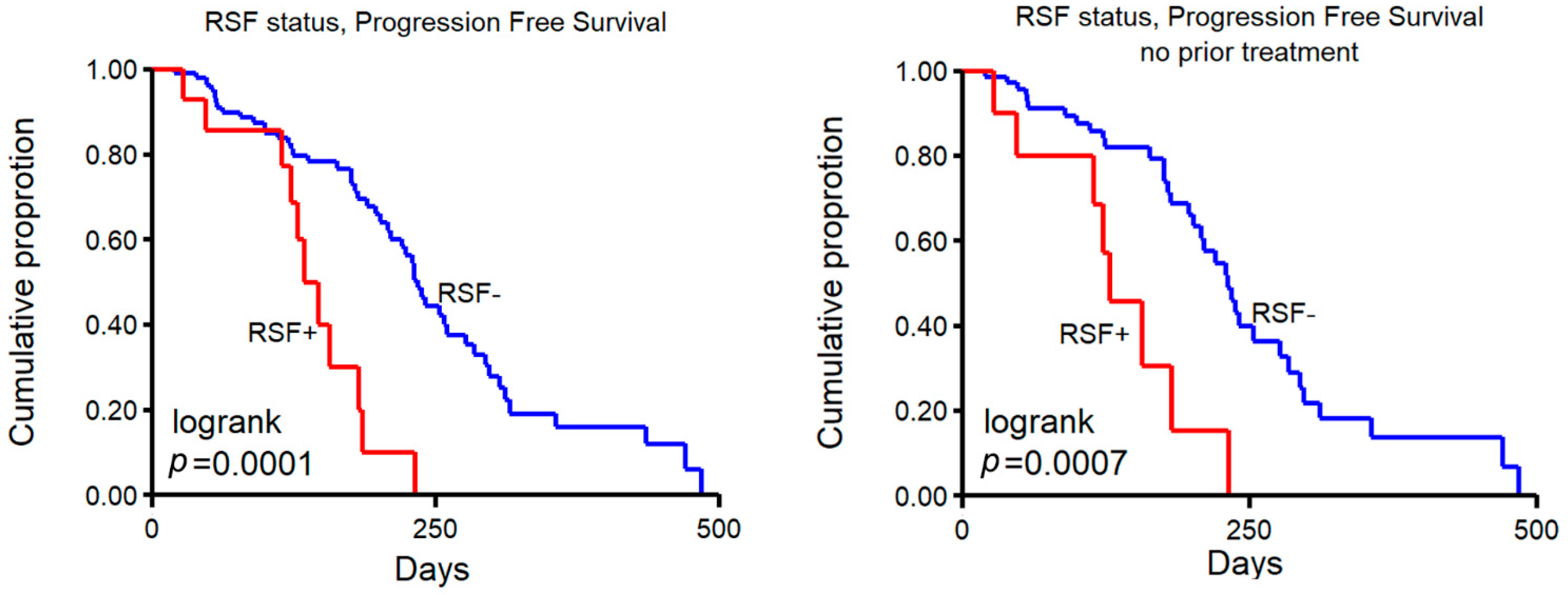
2.4. Clinical Implications
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [Green Version]
- van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.; Van der Roest, B.; Besselink, N.; Janssen, R.; Boymans, S.; Martens, J.W.M.; Yaspo, M.L.; Priestley, P.; Kuijk, E.; Cuppen, E.; et al. 5-Fluorouracil treatment induces characteristic T > G mutations in human cancer. Nat. Commun. 2019, 10, 4571. [Google Scholar] [CrossRef] [Green Version]
- Mendelaar, P.A.J.; Smid, M.; van Riet, J.; Angus, L.; Labots, M.; Steeghs, N.; Hendriks, M.P.; Cirkel, G.A.; van Rooijen, J.M.; Ten Tije, A.J.; et al. Whole genome sequencing of metastatic colorectal cancer reveals prior treatment effects and specific metastasis features. Nat. Commun. 2021, 12, 574. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006, 5, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Nagpal, G.; Kumar, V.; Usmani, S.S.; Agrawal, P.; Raghava, G.P.S. HumCFS: A database of fragile sites in human chromosomes. BMC Genom. 2019, 19, 985. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Langley, A.R.; Graf, S.; Smith, J.C.; Krude, T. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 2016, 44, 10230–10247. [Google Scholar] [CrossRef] [Green Version]
- Boot, A.; Huang, M.N.; Ng, A.W.T.; Ho, S.C.; Lim, J.Q.; Kawakami, Y.; Chayama, K.; Teh, B.T.; Nakagawa, H.; Rozen, S.G. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 2018, 28, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.F.; Le Pla, R.C.; Jones, G.D.; Thomas, A.L.; Farmer, P.B. Determination of cisplatin 1,2-intrastrand guanine-guanine DNA adducts in human leukocytes by high-performance liquid chromatography coupled to inductively coupled plasma mass spectrometry. Chem. Res. Toxicol. 2010, 23, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Scharer, O.D. Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair 2016, 44, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, D.L.; Schroder, J.; Penington, J.S.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.P.; Papenfuss, A.T. GRIDSS: Sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Start Position of First DEL | End Position of Last DEL | Gene | Fragile Site (HumCFS) | Nr. of Samples with >1 DEL |
|---|---|---|---|---|---|
| 1 | 48435860 | 50548351 | AGBL4 | ~0.6 Mb 5′ of FRA1B | 34 |
| 3 | 59572890 | 61536465 | FHIT | FRA3B | 168 |
| 3 | 174058837 | 175755903 | NAALADL2 | ~6.7 Mb 5′ of FRA3C | 80 |
| 4 | 91027502 | 92737250 | CCSER1 | FRA4E | 85 |
| 5 | 58229103 | 59931219 | PDE4D | FRA5H | 55 |
| 6 | 161666892 | 163416771 | PRKN | FRA6E | 125 |
| 7 | 109283013 | 111357245 | IMMP2L | FRA7K | 76 |
| 8 | 89804552 | 91152321 | NBN * | ~3.2 Mb 5′ of FRA8B | 36 |
| 10 | 52569645 | 54085262 | PRKG1 | ~0.6 Mb 5′ of FRA10C | 40 |
| 16 | 5690370 | 7815010 | RBFOX1 | ~7 Mb 5′ of FRA16A | 173 |
| 16 | 78099748 | 79260329 | WWOX | FRA16D | 61 |
| 20 | 13921064 | 16054649 | MACROD2 | ~1.8 Mb 5′ of FRA20A | 207 |
| X | 6511238 | 7782627 | PUDP STS | ~21.9 Mb 5′ of FRAXB | 59 |
| Prior Treatment | Total | RSF+ | RSF- |
|---|---|---|---|
| All | 118 | 14 | 104 |
| Platinum-containing | 18 | 2 | 16 |
| Other | 19 | 2 | 17 |
| None | 81 | 10 | 71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smid, M.; Wilting, S.M.; Martens, J.W.M. Lost by Transcription: Fork Failures, Elevated Expression, and Clinical Consequences Related to Deletions in Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 5080. https://doi.org/10.3390/ijms23095080
Smid M, Wilting SM, Martens JWM. Lost by Transcription: Fork Failures, Elevated Expression, and Clinical Consequences Related to Deletions in Metastatic Colorectal Cancer. International Journal of Molecular Sciences. 2022; 23(9):5080. https://doi.org/10.3390/ijms23095080
Chicago/Turabian StyleSmid, Marcel, Saskia M. Wilting, and John W. M. Martens. 2022. "Lost by Transcription: Fork Failures, Elevated Expression, and Clinical Consequences Related to Deletions in Metastatic Colorectal Cancer" International Journal of Molecular Sciences 23, no. 9: 5080. https://doi.org/10.3390/ijms23095080
APA StyleSmid, M., Wilting, S. M., & Martens, J. W. M. (2022). Lost by Transcription: Fork Failures, Elevated Expression, and Clinical Consequences Related to Deletions in Metastatic Colorectal Cancer. International Journal of Molecular Sciences, 23(9), 5080. https://doi.org/10.3390/ijms23095080