
Ethyl Gallate Dual-Targeting PTPN6 and PPARγ Shows Anti-Diabetic and Anti-Obese Effects

 , ,
, , 
Abstract
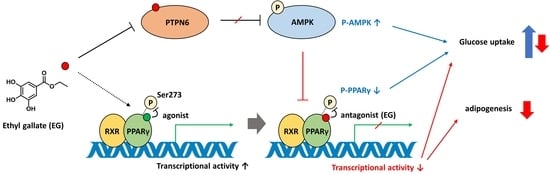
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Depletion of PTPN6 by siRNA Knockdown Increases AMPK Phosphorylation
2.2. The Knockdown of PTPN6 by siRNA Suppresses Adipogenesis of 3T3-L1 Cells
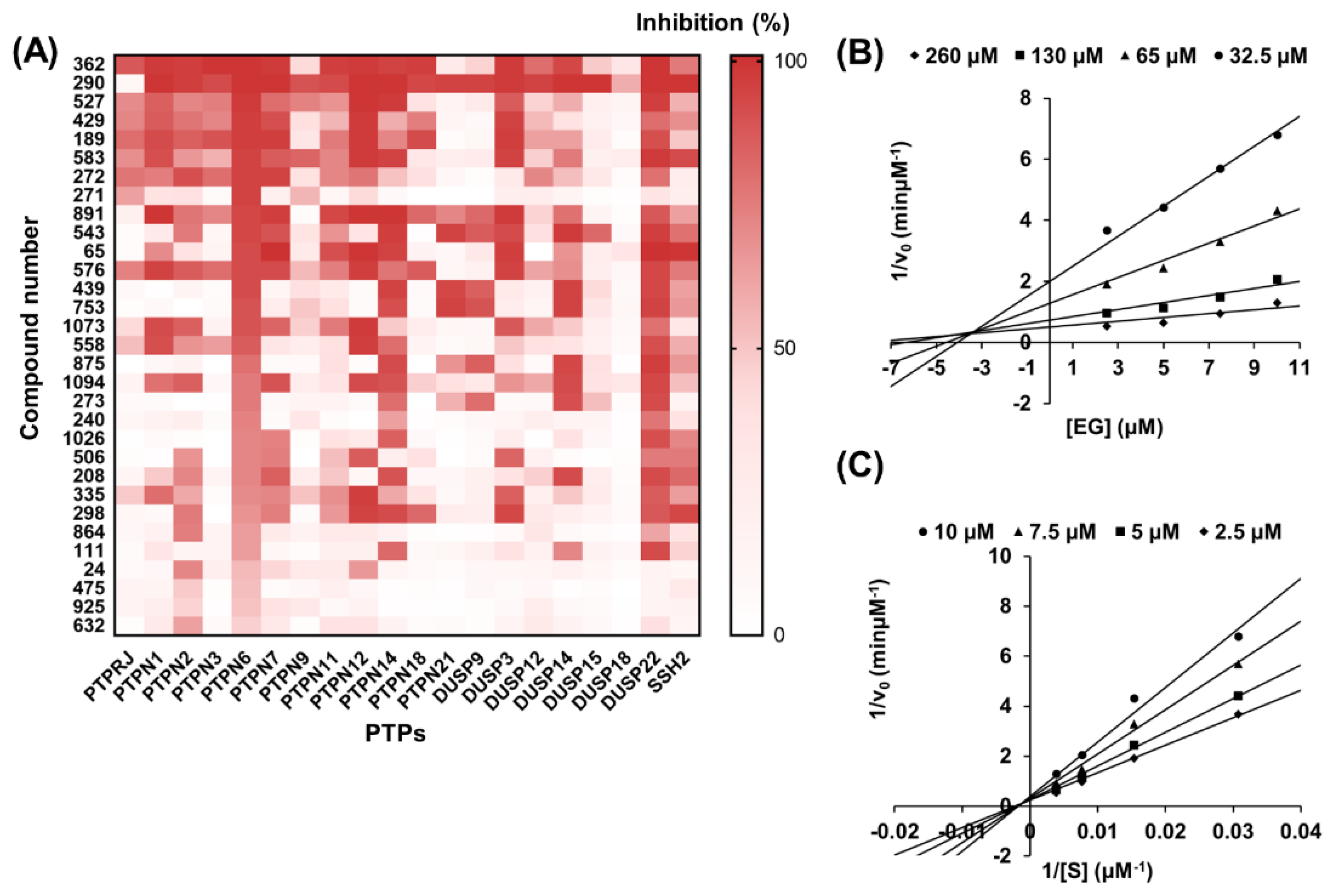
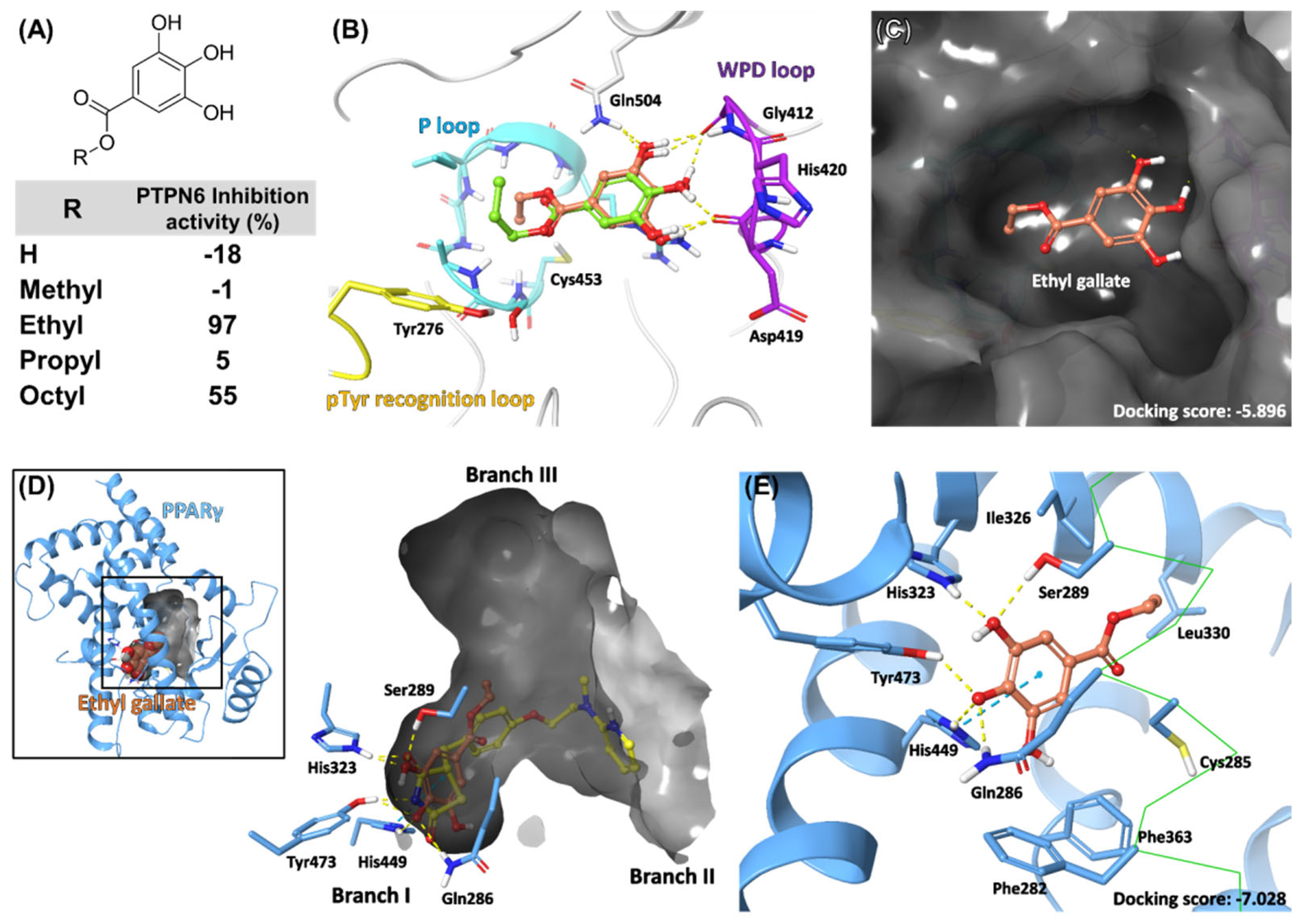
2.3. Selection and Validation of Ethyl Gallate as a PTPN6 Inhibitor
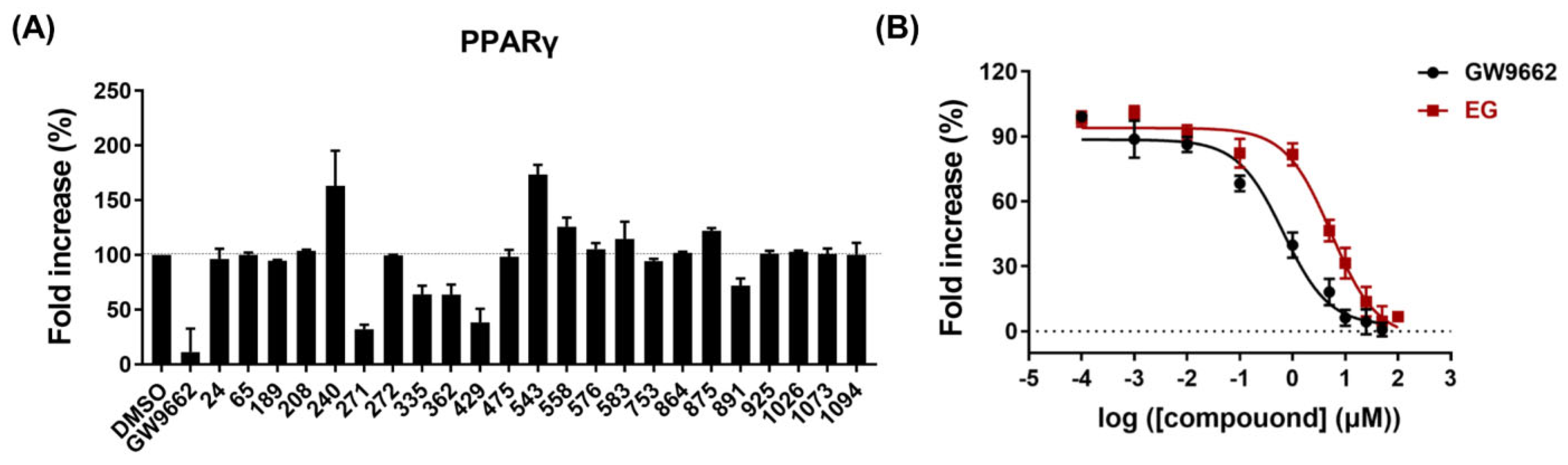
2.4. PPARγ Antagonistic Effect of Ethyl Gallate
2.5. Docking Model of EG on PTPN6 and PPARγ
2.6. EG Treatment Upregulates Glucose Uptake and Activates AMPK in 3T3-L1 Adipocytes
2.7. EG Treatment Suppresses the Early Stage of Adipogenesis in 3T3-L1 Cells
3. Discussion
4. Materials and Methods
4.1. Overexpression and Purification of PTPN6
4.2. Enzymatic Assays
4.3. PPARγ Transactivation Assay
4.4. Docking Study of EG and Gallate Derivatives
4.5. Cell Culture and Differentiation
4.6. RNA Interference
4.7. Quantitative Real Time-Polymerase Chain Reaction (qRT-PCR)
4.8. Glucose Uptake Assay
4.9. Western Blotting
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padwal, R.S.; Sharma, A.M. Prevention of cardiovascular disease: Obesity, diabetes and the metabolic syndrome. Can. J. Cardiol. 2010, 26, 18C–20C. [Google Scholar] [CrossRef] [Green Version]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Farag, Y.M.; Gaballa, M.R. Diabesity: An overview of a rising epidemic. Nephrol Dial. Transplant 2011, 26, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Pappachan, J.M.; Viswanath, A.K. Medical Management of Diabesity: Do We Have Realistic Targets? Curr. Diab. Rep. 2017, 17, 4. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Fernandez, C.J.; Chacko, E.C. Diabesity and antidiabetic drugs. Mol. Aspects Med. 2019, 66, 3–12. [Google Scholar] [CrossRef]
- Brown, E.; Heerspink, H.J.L.; Cuthbertson, D.J.; Wilding, J.P.H. SGLT2 inhibitors and GLP-1 receptor agonists: Established and emerging indications. Lancet 2021, 398, 262–276. [Google Scholar] [CrossRef]
- Brusotti, G.; Montanari, R.; Capelli, D.; Cattaneo, G.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Peiretti, F.; Bonardo, B.; Paiardini, A.; et al. Betulinic acid is a PPARgamma antagonist that improves glucose uptake, promotes osteogenesis and inhibits adipogenesis. Sci. Rep. 2017, 7, 5777. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dai, L.; Chen, J. Identification of a proliferator-activated receptor-gamma antagonist for the treatment of type 2 diabetes mellitus. Exp. Ther. Med. 2015, 9, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.S.; Kim, E.S.; Jung, J.E.; Marciano, D.P.; Jo, A.; Koo, J.Y.; Choi, S.Y.; Yang, Y.R.; Jang, H.J.; Kim, E.K.; et al. PPARgamma Antagonist Gleevec Improves Insulin Sensitivity and Promotes the Browning of White Adipose Tissue. Diabetes 2016, 65, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Frkic, R.L.; Marshall, A.C.; Blayo, A.L.; Pukala, T.L.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. PPARgamma in Complex with an Antagonist and Inverse Agonist: A Tumble and Trap Mechanism of the Activation Helix. iScience 2018, 5, 69–79. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef]
- Frkic, R.L.; Richter, K.; Bruning, J.B. The therapeutic potential of inhibiting PPARgamma phosphorylation to treat type 2 diabetes. J. Biol. Chem. 2021, 297, 101030. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Li, Y.; Zhao, T.; Wang, Y.; Sun, C. Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through LKB1/AMPK pathway. PLoS ONE 2013, 8, e70135. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Lee, W.S.; Hwang, J.T.; Kwon, D.Y.; Surh, Y.J.; Park, O.J. Curcumin exerts antidifferentiation effect through AMPKalpha-PPAR-gamma in 3T3-L1 adipocytes and antiproliferatory effect through AMPKalpha-COX-2 in cancer cells. J. Agric. Food Chem. 2009, 57, 305–310. [Google Scholar] [CrossRef]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Pu, R.; Shi, D.; Gan, T.; Ren, X.; Ba, Y.; Huo, Y.; Bai, Y.; Zheng, T.; Cheng, N. Effects of metformin in obesity treatment in different populations: A meta-analysis. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820926000. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases-from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.Y.; Kang, H.J.; Ahn, D.; Hwang, J.Y.; Kwon, S.J.; Chung, S.J. Identification of chebulinic acid as a dual targeting inhibitor of protein tyrosine phosphatases relevant to insulin resistance. Bioorg. Chem. 2019, 90, 103087. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Lee, J.H.; Kwon, S.J.; Kang, H.J.; Chung, S.J. Ginkgolic acid as a dual-targeting inhibitor for protein tyrosine phosphatases relevant to insulin resistance. Bioorg. Chem. 2018, 81, 264–269. [Google Scholar] [CrossRef]
- Lin, L.; Jian, J.; Song, C.Y.; Chen, F.; Ding, K.; Xie, W.F.; Hu, P.F. SHP-1 ameliorates nonalcoholic steatohepatitis by inhibiting proinflammatory cytokine production. FEBS Lett. 2020, 594, 2965–2974. [Google Scholar] [CrossRef]
- Sharma, Y.; Ahmad, A.; Yavvari, P.S.; Kumar Muwal, S.; Bajaj, A.; Khan, F. Targeted SHP-1 Silencing Modulates the Macrophage Phenotype, Leading to Metabolic Improvement in Dietary Obese Mice. Mol. Ther. Nucleic Acids. 2019, 16, 626–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, E.; Forest, M.P.; Schwab, M.; Avramoglu, R.K.; St-Amand, E.; Caron, A.Z.; Bellmann, K.; Shum, M.; Voisin, G.; Paquet, M.; et al. Hepatocyte-specific Ptpn6 deletion promotes hepatic lipid accretion, but reduces NAFLD in diet-induced obesity: Potential role of PPARgamma. Hepatology 2014, 59, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Laqui-Estana, J.; Lopez-Solis, R.; Pena-Neira, A.; Medel-Maraboli, M.; Obreque-Slier, E. Wines in contact with oak wood: The impact of the variety (Carmenere and Cabernet Sauvignon), format (barrels, chips and staves), and aging time on the phenolic composition. J. Sci. Food Agric. 2019, 99, 436–448. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.H.S.; Simas, N.K.; Alviano, C.S.; Alviano, D.S.; Ventura, J.A.; de Lima, E.J.; Seabra, S.H.; Kuster, R.M. Anti-Escherichia coli activity of extracts from Schinus terebinthifolius fruits and leaves. Nat. Prod. Res. 2018, 32, 1365–1368. [Google Scholar] [CrossRef]
- Lee, J.; Chan, B.L.; Mitchell, A.E. Identification/quantification of free and bound phenolic acids in peel and pulp of apples (Malus domestica) using high resolution mass spectrometry (HRMS). Food Chem. 2017, 215, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liao, L.; Moore, J.; Wu, T.; Wang, Z. Antioxidant phenolic compounds from walnut kernels (Juglans regia L.). Food Chem. 2009, 113, 160–165. [Google Scholar] [CrossRef]
- Kalaivani, T.; Rajasekaran, C.; Mathew, L. Free radical scavenging, cytotoxic, and hemolytic activities of an active antioxidant compound ethyl gallate from leaves of Acacia nilotica (L.) Wild. Ex. Delile subsp. indica (Benth.) Brenan. J. Food Sci. 2011, 76, T144–T149. [Google Scholar] [CrossRef]
- Cui, H.; Yuan, J.; Du, X.; Wang, M.; Yue, L.; Liu, J. Ethyl gallate suppresses proliferation and invasion in human breast cancer cells via Akt-NF-kappaB signaling. Oncol. Rep. 2015, 33, 1284–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehla, K.; Balwani, S.; Kulshreshtha, A.; Nandi, D.; Jaisankar, P.; Ghosh, B. Ethyl gallate isolated from Pistacia integerrima Linn. inhibits cell adhesion molecules by blocking AP-1 transcription factor. J. Ethnopharmacol. 2011, 137, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, J.; Xing, S.; Pan, X.; Wei, S.; Zhou, M.; Li, Z.; Wang, L.; Bielicki, J.K. The benzoate plant metabolite ethyl gallate prevents cellular- and vascular-lipid accumulation in experimental models of atherosclerosis. Biochem. Biophys. Res. Commun. 2021, 556, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, Y.; Ahn, D.; Chung, S.J. Protein tyrosine phosphatases (PTPs) in diabetes: Causes and therapeutic opportunities. Arch. Pharm. Res. 2021, 44, 310–321. [Google Scholar] [CrossRef]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Muller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Yang, S.; Wakabayashi, K.; Svensson, M.N.D.; Stanford, S.M.; Santelli, E.; Bottini, N. RPTPalpha phosphatase activity is allosterically regulated by the membrane-distal catalytic domain. J. Biol. Chem. 2020, 295, 4923–4936. [Google Scholar] [CrossRef]
- Wang, W.; Liu, L.; Song, X.; Mo, Y.; Komma, C.; Bellamy, H.D.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J. Cell Biochem. 2011, 112, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, L.; He, D.; Song, X.; Liang, X.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 2003, 278, 6516–6520. [Google Scholar] [CrossRef] [Green Version]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARgamma. Elife 2018, 7, e43320. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Furukawa, N.; Escaron, P.; Weiszmann, J.; Lee, G.; Lindstrom, M.; Liu, J.; Liu, X.; Xu, H.; et al. T2384, a novel antidiabetic agent with unique peroxisome proliferator-activated receptor gamma binding properties. J. Biol. Chem. 2008, 283, 9168–9176. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.L.; Min, J.K.; Roh, K.M.; Kim, W.K.; Han, B.S.; Bae, K.H.; Lee, S.C.; Chung, S.J.; Kang, H.J. Phosphoprotein phosphatase 1CB (PPP1CB), a novel adipogenic activator, promotes 3T3-L1 adipogenesis. Biochem. Biophys. Res. Commun. 2015, 467, 211–217. [Google Scholar] [CrossRef]
- Doan, K.V.; Ko, C.M.; Kinyua, A.W.; Yang, D.J.; Choi, Y.H.; Oh, I.Y.; Nguyen, N.M.; Ko, A.; Choi, J.W.; Jeong, Y.; et al. Gallic acid regulates body weight and glucose homeostasis through AMPK activation. Endocrinology 2015, 156, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.K.; Lee, J.; Hong, V.S.; Choi, J.S.; Lee, T.Y.; Jang, B.C. Identification of KMU-3, a novel derivative of gallic acid, as an inhibitor of adipogenesis. PLoS ONE 2014, 9, e109344. [Google Scholar]
- Yuan, X.; Bu, H.; Zhou, J.; Yang, C.Y.; Zhang, H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J. Med. Chem. 2020, 63, 11368–11396. [Google Scholar] [CrossRef]
- Chang, E.; Kim, C.Y. Natural Products and Obesity: A Focus on the Regulation of Mitotic Clonal Expansion during Adipogenesis. Molecules 2019, 24, 1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakab, J.; Miskic, B.; Miksic, S.; Juranic, B.; Cosic, V.; Schwarz, D.; Vcev, A. Adipogenesis as a Potential Anti-Obesity Target: A Review of Pharmacological Treatment and Natural Products. Diabetes Metab. Syndr. Obes. 2021, 14, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Feng, S.; Reuss, L.; Wang, Y. Potential of Natural Products in the Inhibition of Adipogenesis through Regulation of PPARgamma Expression and/or Its Transcriptional Activity. Molecules 2016, 21, 1278. [Google Scholar] [CrossRef] [PubMed]
- Weiszenstein, M.; Musutova, M.; Plihalova, A.; Westlake, K.; Elkalaf, M.; Koc, M.; Prochazka, A.; Pala, J.; Gulati, S.; Trnka, J.; et al. Adipogenesis, lipogenesis and lipolysis is stimulated by mild but not severe hypoxia in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2016, 478, 727–732. [Google Scholar] [CrossRef]
- Arner, P.; Langin, D. Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends Endocrinol. Metab. 2014, 25, 255–262. [Google Scholar] [CrossRef]
- Green, H.; Meuth, M. An established pre-adipose cell line and its differentiation in culture. Cell 1974, 3, 127–133. [Google Scholar] [CrossRef]
- Guru, A.; Issac, P.K.; Velayutham, M.; Saraswathi, N.T.; Arshad, A.; Arockiaraj, J. Molecular mechanism of down-regulating adipogenic transcription factors in 3T3-L1 adipocyte cells by bioactive anti-adipogenic compounds. Mol. Biol. Rep. 2021, 48, 743–761. [Google Scholar] [CrossRef]
- Takahashi, M.; Takahashi, Y.; Takahashi, K.; Zolotaryov, F.N.; Hong, K.S.; Kitazawa, R.; Iida, K.; Okimura, Y.; Kaji, H.; Kitazawa, S.; et al. Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett. 2008, 582, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Ahn, D.; Chung, S.J. Chebulinic Acid Suppresses Adipogenesis in 3T3-L1 Preadipocytes by Inhibiting PPP1CB Activity. Int. J. Mol. Sci. 2022, 23, 865. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Ahn, D.; Hwang, J.Y.; Kang, M.J.; Chung, S.J. Linoleic acid exerts antidiabetic effects by inhibiting protein tyrosine phosphatases associated with insulin resistance. J. Funct. Foods 2021, 83, 104532. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, D.; Kim, J.; Nam, G.; Zhao, X.; Kwon, J.; Hwang, J.Y.; Kim, J.K.; Yoon, S.-Y.; Chung, S.J. Ethyl Gallate Dual-Targeting PTPN6 and PPARγ Shows Anti-Diabetic and Anti-Obese Effects. Int. J. Mol. Sci. 2022, 23, 5020. https://doi.org/10.3390/ijms23095020
Ahn D, Kim J, Nam G, Zhao X, Kwon J, Hwang JY, Kim JK, Yoon S-Y, Chung SJ. Ethyl Gallate Dual-Targeting PTPN6 and PPARγ Shows Anti-Diabetic and Anti-Obese Effects. International Journal of Molecular Sciences. 2022; 23(9):5020. https://doi.org/10.3390/ijms23095020
Chicago/Turabian StyleAhn, Dohee, Jinsoo Kim, Gibeom Nam, Xiaodi Zhao, Jihee Kwon, Ji Young Hwang, Jae Kwan Kim, Sun-Young Yoon, and Sang J. Chung. 2022. "Ethyl Gallate Dual-Targeting PTPN6 and PPARγ Shows Anti-Diabetic and Anti-Obese Effects" International Journal of Molecular Sciences 23, no. 9: 5020. https://doi.org/10.3390/ijms23095020
APA StyleAhn, D., Kim, J., Nam, G., Zhao, X., Kwon, J., Hwang, J. Y., Kim, J. K., Yoon, S.-Y., & Chung, S. J. (2022). Ethyl Gallate Dual-Targeting PTPN6 and PPARγ Shows Anti-Diabetic and Anti-Obese Effects. International Journal of Molecular Sciences, 23(9), 5020. https://doi.org/10.3390/ijms23095020