
CfDNA Measurement as a Diagnostic Tool for the Detection of Brain Somatic Mutations in Refractory Epilepsy

 , , , and
, , , and
Abstract
:1. Introduction
2. Discussion
2.1. Somatic Variants in the Brain and Refractory Epilepsies
2.2. Somatic Variants in CSF CfDNA and Refractory Epilepsy
2.2.1. Cell-Free DNA
2.2.2. Somatic Mutations in CSF and Epilepsy
2.3. Future Perspectives: Somatic Brain Variants Detection in Plasma CfDNA
2.3.1. Blood-Brain Barrier Integrity and Epitoy
2.3.2. Requirements for Specific Brain CfDNA Measurement in Plasma from Patients with Refractory Epileptic
2.3.3. Treatment
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ASD | Autism spectrum disorders |
| BBB | Blood-brain barrier |
| CDG2M | Congenital disorder of glycosylation type IIm |
| cfDNA | Cell-free desoxyribonucleic acid |
| CNS | Central nervous system |
| CNV | Copy Number Variants |
| CSF | Cerebrospinal fluid |
| ddPCR | Droplet digital polymerase chain reaction |
| EE | Epileptic encephalopathies |
| FCD | Focal cortical dysplasia |
| FCDII | Focal cortical dysplasia type II |
| gDNA | Genomic desoxyribonucleic acid |
| HME | hemimegalencephaly |
| ID | Intellectual disability |
| IGE | Idiopathic generalized epilepsy |
| KD | Ketogenic diet |
| LOH | Loss-of-heterozygosity |
| MCD | Malformations of cortical development |
| MOGHE | Mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy |
| NGS | Next-generation sequencing |
| RE | Refractory epilepsy |
| Shh | Sonic hedgehog |
| SNV | Single Nucleotide Variants |
| SWS | Sturge-Weber syndrome |
| TLE | Temporal lobe epilepsy |
| XTLE | Extra temporal lobe epilepsy |
Appendix A

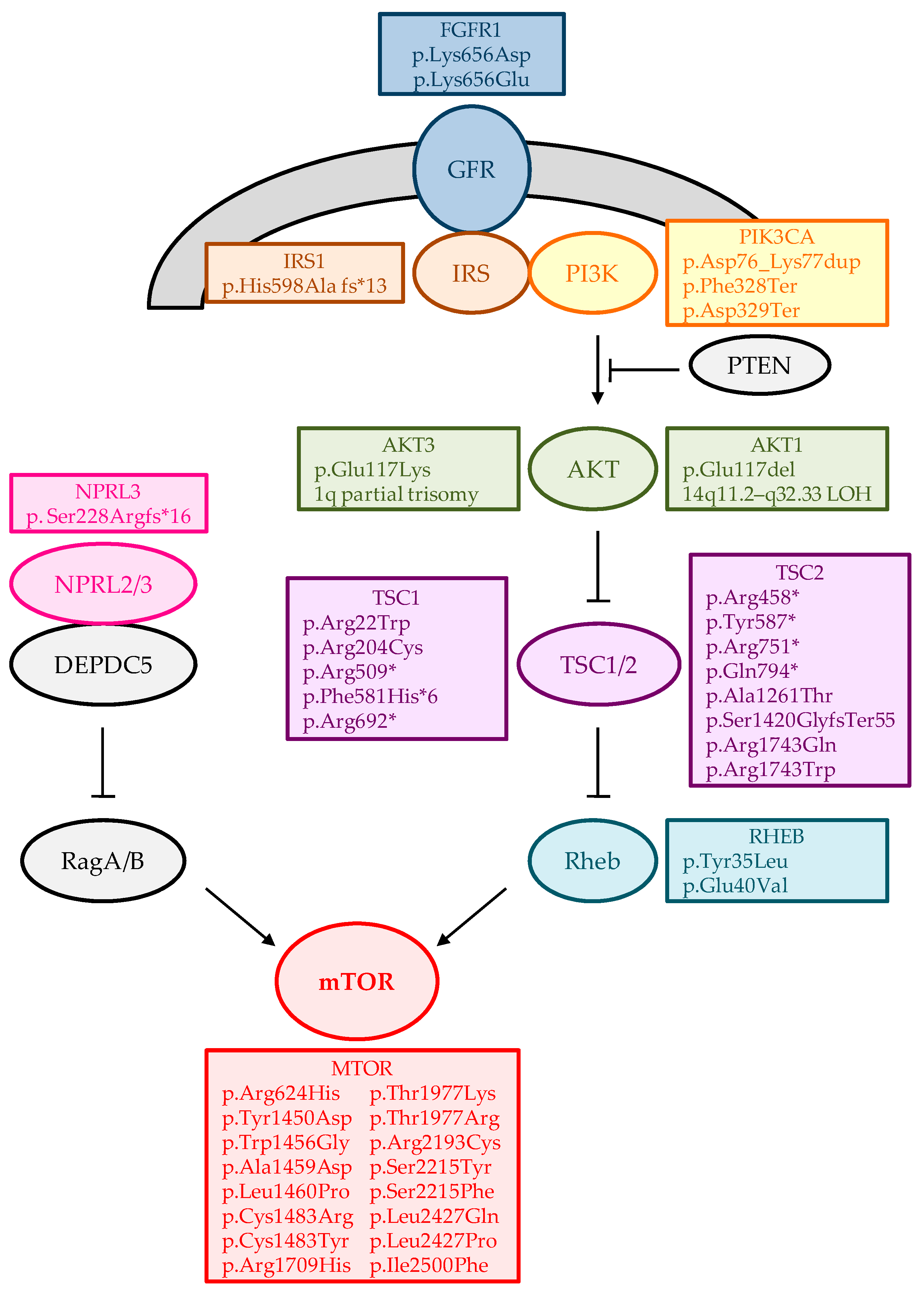{kind=link}
| Analytical Type | Targets | Applications | Technique | Sensitivity (LoD) | Advantages | Limitations |
|---|---|---|---|---|---|---|
| Targeted | Known and unknown mutations, indels, CNV, chromosomal rearrangements (capture) | Cancer detection and monitoring classification, targetable alterations, for research use | Tam-Seq | 2% | High specificity | Amplicon methods by multiplex PCR (depend on fragment size), no error correction |
| eTam-Seq | 0.02% | Error correction | Amplicon methods by multiplex PCR | |||
| Safe-SeqS | 0.01–0.05% | Error correction by SSCS | Amplicon methods by multiplex PCR | |||
| Duplex Sequencing | 0.0001–0.1% | Error correction by DSCS | Amplicon methods by multiplex PCR | |||
| TEC-Seq | 0.05–0.1% | Error correction by SSCS, Hybrid capture method (not dependent on fragment size) | Less comprehensive than WGS or WES | |||
| single primer extension (SPE) | 0.5–1% | Amplicon methods by SPE (not dependent on fragment size), error correction by SSCS | Less comprehensive than WGS or WES | |||
| SPE-duplex UMI | 0.1–0.2% | Error correction by DSCS | Less comprehensive than WGS or WES | |||
| CAPP-Seq | 0.02% | Hybrid capture method (not dependent on fragment size) | Need large input, allelic bias (capture), stereotypical errors(hybridization step), less comprehensive than WGS or WES | |||
| iDES eCAPP-Seq | 0.00025–0.004% | Error correction by DSCS and correction of stereotypical errors | Less comprehensive than WGS or WES | |||
| VDJ rearrangements | Non-invasive monitoring, approved for clinical use | Ig-HTS | 0.001% | Very high sensitivity | Tissue biopsy needed | |
| Untargeted | Coding regions, intron-exon junctions, promoters, untranslated regions, non-coding DNA of miRNA genes | Monogenic disorders. Cancer detection, monitoring of resistant clones in metastasis, for research use | WES | 5% | Mutation discovery and signatures, detection of CNV, fusion genes, rearrangements, predicted neoantigens, and mutational burden. | Low sensitivity (increasing depth leads to high cost) and need of bioinformatic expertise. |
| Aneuploidies. Structural variants (fragmentation pattern, genome-wide CNV, methylation profile) | Non-invasive prenatal diagnosis for clinical use. cancer localization and origin, and early detection (early and late-stage) | WGS | 5–10% | Shallow sequencing, genome-wide profiling, identification of cancer signatures | Expensive, variable sensitivity (low) and specificity, need bioinformatics expertise, lots of data generated. |
References
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug resistance in epilepsy: Clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshe, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- Hauser, R.M.; Henshall, D.C.; Lubin, F.D. The Epigenetics of Epilepsy and Its Progression. Neuroscientist 2018, 24, 186–200. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; Dahl, H.-H.M.; Damiano, J.A.; Smith, R.J.H.; Scheffer, I.E.; Berkovic, S.F. Recent advances in the molecular genetics of epilepsy. J. Med. Genet. 2013, 50, 271–279. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 2018, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The Genetics of Epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef]
- Battaglia, A.; Guerrini, R. Chromosomal disorders associated with epilepsy. Epileptic Disord. 2005, 7, 181–192. [Google Scholar]
- Schinzel, A. Catalogue of Unbalanced Aberrations in Man; De Gruyter: Berlin, Germany, 2001; ISBN 3110116073. [Google Scholar]
- Mayo, S. Search, and identification of new epigenetic or genetic causes of neurodevelopmental disorders. Univ. Valencia 2015. Available online: http://hdl.handle.net/10550/47942 (accessed on 25 April 2022).
- Rosello, M.; Martinez, F.; Monfort, S.; Mayo, S.; Oltra, S.; Orellana, C. Phenotype profiling of patients with intellectual disability and copy number variations. Eur. J. Paediatr. Neurol. 2014, 18, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Campbell, C.D.; Eichler, E.E. Human copy number variation and complex genetic disease. Annu. Rev. Genet. 2011, 45, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Yendle, S.C.; Hsu, C.; Cook, J.; Geraghty, E.; McMahon, J.M.; Eeg-Olofsson, O.; Sadleir, L.G.; Gill, D.; Ben-Zeev, B.; et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 2011, 70, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northrup, H.; Koenig, M.K.; Pearson, D.A.; Au, K.S. Tuberous Sclerosis Complex; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Claes, L.; Del-Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, X.; Li, J.; Shao, Q.; Chen, H.; Lin, Z.; Sun, Z.S.; Wu, J. EpilepsyGene: A genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res. 2015, 43, D893–D899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lin, Z.-J.; Liu, L.; Xu, H.-Q.; Shi, Y.-W.; Yi, Y.-H.; He, N.; Liao, W.-P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Helbig, K.L.; Farwell Hagman, K.D.; Shinde, D.N.; Mroske, C.; Powis, Z.; Li, S.; Tang, S.; Helbig, I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet. Med. 2016, 18, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Pang, N.; Wang, Y.; Wang, X.-L.; Chen, J.; Xiong, J.; Peng, P.; Zhu, C.-H.; Kessi, M.B.; He, F.; et al. Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci. Ther. 2018, 25, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.F.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef]
- Miller, I.O.; Sotero De Menezes, M.A. SCN1A Seizure Disorders. In GeneReviews; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Stouffer, M.A.; Golden, J.A.; Francis, F. Neuronal migration disorders: Focus on the cytoskeleton and epilepsy. Neurobiol. Dis. 2016, 92, 18–45. [Google Scholar] [CrossRef] [PubMed]
- Poduri, A.; Evrony, G.D.; Cai, X.; Walsh, C.A. Somatic mutation, genomic variation, and neurological disease. Science 2013, 341, 1237758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.; McQuillan, L.; Poduri, A.; Green, T.E.; Matsumoto, N.; Mefford, H.C.; Scheffer, I.E.; Berkovic, S.F.; Hildebrand, M.S. Somatic mutation: The hidden genetics of brain malformations and focal epilepsies. Epilepsy Res. 2019, 155, 106161. [Google Scholar] [CrossRef] [PubMed]
- Rodin, R.E.; Walsh, C.A. Somatic Mutation in Pediatric Neurological Diseases. Pediatr. Neurol. 2018, 87, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Bruno, D.C.F.; Donatti, A.; Martin, M.; Almeida, V.S.; Geraldis, J.C.; Oliveira, F.S.; Dogini, D.B.; Lopes-Cendes, I. Circulating nucleic acids in the plasma and serum as potential biomarkers in neurological disorders. Braz. J. Med. Biol. Res. 2020, 53, e9881. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, L.; Dibbens, L.M.; Lawrence, K.M.; Iona, X.; McMahon, J.M.; Murrell, W.; Mackay-Sim, A.; Scheffer, I.E.; Berkovic, S.F. Timing of de novo mutagenesis–A twin study of sodium-channel mutations. N. Engl. J. Med. 2010, 363, 1335–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, M.J.; Moran, J.V.; Abyzov, A.; Akbarian, S.; Bae, T.; Cortes-Ciriano, I.; Erwin, J.A.; Fasching, L.; Flasch, D.A.; Freed, D.; et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 2017, 356, eaal1641. [Google Scholar] [CrossRef] [Green Version]
- Rehen, S.K.; Yung, Y.C.; McCreight, M.P.; Kaushal, D.; Yang, A.H.; Almeida, B.S.V.; Kingsbury, M.A.; Cabral, K.M.S.; McConnell, M.J.; Anliker, B.; et al. Constitutional aneuploidy in the normal human brain. J. Neurosci. 2005, 25, 2176–2180. [Google Scholar] [CrossRef]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Chatterton, Z.; Pflueger, J.; Damiano, J.A.; McQuillan, L.; Harvey, A.S.; Malone, S.; Do, H.; Maixner, W.; Schneider, A.; et al. Cerebrospinal fluid liquid biopsy for detecting somatic mosaicism in brain. Brain Commun. 2021, 3, fcaa235. [Google Scholar] [CrossRef]
- Avansini, S.H.; Torres, F.R.; Vieira, A.S.; Dogini, D.B.; Rogerio, F.; Coan, A.C.; Morita, M.E.; Guerreiro, M.M.; Yasuda, C.L.; Secolin, R.; et al. Dysregulation of NEUROG2 plays a key role in focal cortical dysplasia. Ann. Neurol. 2018, 83, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, M.S.; Griffin, N.G.; Damiano, J.A.; Cops, E.J.; Burgess, R.; Ozturk, E.; Jones, N.C.; Leventer, R.J.; Freeman, J.L.; Harvey, A.S.; et al. Mutations of the Sonic Hedgehog Pathway Underlie Hypothalamic Hamartoma with Gelastic Epilepsy. Am. J. Hum. Genet. 2016, 99, 423–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, V.; Pantaleo, M.; Barba, C.; Baroni, G.; Mei, D.; Buccoliero, A.M.; Giglio, S.; Giordano, F.; Baek, S.T.; Gleeson, J.G.; et al. Focal dysplasia of the cerebral cortex and infantile spasms associated with somatic 1q21.1-q44 duplication including the AKT3 gene. Clin. Genet. 2015, 88, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Poduri, A.; Evrony, G.D.; Cai, X.; Elhosary, P.C.; Beroukhim, R.; Lehtinen, M.K.; Hills, L.B.; Heinzen, E.L.; Hill, R.S.; Barry, B.J.; et al. Somatic Activation of AKT3 Causes Hemispheric Developmental Brain Malformations. Neuron 2012, 74, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef] [Green Version]
- D’Gama, A.M.; Woodworth, M.B.; Hossain, A.A.; Bizzotto, S.; Hatem, N.E.; LaCoursiere, C.M.; Najm, I.; Ying, Z.; Yang, E.; Barkovich, A.J.; et al. Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep. 2017, 21, 3754–3766. [Google Scholar] [CrossRef] [Green Version]
- Baldassari, S.; Ribierre, T.; Marsan, E.; Adle-Biassette, H.; Ferrand-Sorbets, S.; Bulteau, C.; Dorison, N.; Fohlen, M.; Polivka, M.; Weckhuysen, S.; et al. Dissecting the genetic basis of focal cortical dysplasia: A large cohort study. Acta Neuropathol. 2019, 138, 885–900. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.S.; Ko, A.; Kim, W.K.; Kim, S.H.; Kim, J.S.; Shim, K.W.; Aronica, E.; Mijnsbergen, C.; Spliet, W.G.M.; Koh, H.Y.; et al. Precise detection of low-level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol. 2019, 138, 901–912. [Google Scholar] [CrossRef]
- Blümcke, I.; Coras, R.; Busch, R.M.; Morita-Sherman, M.; Lal, D.; Prayson, R.; Cendes, F.; Lopes-Cendes, I.; Rogerio, F.; Almeida, V.S.; et al. Toward a better definition of focal cortical dysplasia: An iterative histopathological and genetic agreement trial. Epilepsia 2021, 62, 1416–1428. [Google Scholar] [CrossRef]
- Niestroj, L.M.; May, P.; Artomov, M.; Kobow, K.; Coras, R.; Pérez-Palma, E.; Altmüller, J.; Thiele, H.; Nürnberg, P.; Leu, C.; et al. Assessment of genetic variant burden in epilepsy-associated brain lesions. Eur. J. Hum. Genet. 2019, 27, 1738–1744. [Google Scholar] [CrossRef]
- Kim, S.; Baldassari, S.; Sim, N.S.; Chipaux, M.; Dorfmüller, G.; Kim, D.S.; Chang, W.S.; Taly, V.; Lee, J.H.; Baulac, S. Detection of Brain Somatic Mutations in Cerebrospinal Fluid from Refractory Epilepsy Patients. Ann. Neurol. 2021, 89, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Harvey, A.S.; Malone, S.; Damiano, J.A.; Do, H.; Ye, Z.; McQuillan, L.; Maixner, W.; Kalnins, R.; Nolan, B.; et al. Somatic GNAQ mutation in the forme fruste of Sturge-Weber syndrome. Neurol. Genet. 2018, 4, e236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Gao, K.; Liu, Q.; Zhou, J.; Li, X.; Lang, N.; Liu, M.; Wang, T.; Zhang, J.; Wang, H.; et al. Somatic variants in new candidate genes identified in focal cortical dysplasia type II. Epilepsia 2020, 61, 667–678. [Google Scholar] [CrossRef]
- Von Wrede, R.; Jeub, M.; Ariöz, I.; Elger, C.E.; von Voss, H.; Klein, H.G.; Becker, A.J.; Schoch, S.; Surges, R.; Kunz, W.S. Novel kcnh1 mutations associated with epilepsy: Broadening the phenotypic spectrum of kcnh1-associated diseases. Genes 2021, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Kim, W.I.; Kang, H.C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.W.; Kim, S.; Kim, H.M.; Kim, J.A.; et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 2015, 21, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Leventer, R.J.; Scerri, T.; Marsh, A.P.L.; Pope, K.; Gillies, G.; Maixner, W.; MacGregor, D.; Harvey, A.S.; Delatycki, M.B.; Amor, D.J.; et al. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurology 2015, 84, 2029–2032. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Stephenson, S.E.M.; Pope, K.; Gillies, G.; Maixner, W.; Macdonald-laurs, E.; MacGregor, D.; D’Arcy, C.; Jackson, G.; Harvey, A.S.; et al. Genetic Characterization Identifies Bottom-of-Sulcus Dysplasia as an mTORopathy. Neurology 2020, 95, e2542–e2551. [Google Scholar] [CrossRef]
- Hanai, S.; Sukigara, S.; Dai, H.; Owa, T.; Horike, S.I.; Otsuki, T.; Saito, T.; Nakagawa, E.; Ikegaya, N.; Kaido, T.; et al. Pathologic Active mTOR Mutation in Brain Malformation with Intractable Epilepsy Leads to Cell-Autonomous Migration Delay. Am. J. Pathol. 2017, 187, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Szczałuba, K.; Rydzanicz, M.; Walczak, A.; Kosińska, J.; Koppolu, A.; Biernacka, A.; Iwanicka-Pronicka, K.; Grajkowska, W.; Jurkiewicz, E.; Kowalczyk, P.; et al. Brain tissue low-level mosaicism for mtor mutation causes smith–kingsmore phenotype with recurrent hypoglycemia—a novel phenotype and a further proof for testing of an affected tissue. Diagnostics 2021, 11, 1269. [Google Scholar] [CrossRef]
- Salinas, V.; Vega, P.; Piccirilli, M.V.; Chicco, C.; Ciraolo, C.; Christiansen, S.; Consalvo, D.; Perez-Maturo, J.; Medina, N.; González-Morón, D.; et al. Identification of a somatic mutation in the RHEB gene through high depth and ultra-high depth next generation sequencing in a patient with Hemimegalencephaly and drug resistant Epilepsy. Eur. J. Med. Genet. 2019, 62, 103571. [Google Scholar] [CrossRef]
- Bonduelle, T.; Hartlieb, T.; Baldassari, S.; Sim, N.S.; Kim, S.H.; Kang, H.C.; Kobow, K.; Coras, R.; Chipaux, M.; Dorfmüller, G.; et al. Frequent SLC35A2 brain mosaicism in mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE). Acta Neuropathol. Commun. 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.S.; Seo, Y.; Lim, J.S.; Kim, W.K.; Son, H.; Kim, H.D.; Kim, S.; An, H.J.; Kang, H.C.; Kim, S.H.; et al. Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurol. Genet. 2018, 4, e294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winawer, M.R.; Griffin, N.G.; Samanamud, J.; Baugh, E.H.; Rathakrishnan, D.; Ramalingam, S.; Zagzag, D.; Schevon, C.A.; Dugan, P.; Hegde, M.; et al. Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann. Neurol. 2018, 83, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Koboldt, D.C.; Schieffer, K.M.; Bedrosian, T.A.; Crist, E.; Sheline, A.; Leraas, K.; Magrini, V.; Zhong, H.; Brennan, P.; et al. Somatic SLC35A2 mosaicism correlates with clinical findings in epilepsy brain tissue. Neurol. Genet. 2020, 6, e460. [Google Scholar] [CrossRef]
- Jha, R.; Kurup, A.; Kovilapu, U.B.; Ranjan, R.; Sondhi, V. Somatic mutations involving TSC 1 and TSC2 genes in two children with focal cortical dysplasia. Brain Dev. 2022, 44, 166–172. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; de Sampaio e Spohr, T.C.L. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Lipton, J.O.; Sahin, M. The Neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Aronica, E.; Becker, A.J.; Spreafico, R. Malformations of cortical development. Brain Pathol. 2012, 22, 380–401. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, J.H. Mechanistic target of rapamycin pathway in epileptic disorders. J. Korean Neurosurg. Soc. 2019, 62, 272–287. [Google Scholar] [CrossRef]
- Lim, J.S.; Lee, J.H. Brain somatic mutations in MTOR leading to focal cortical dysplasia. BMB Rep. 2016, 49, 71–72. [Google Scholar] [CrossRef] [Green Version]
- Stroun, M.; Maurice, P.; Vasioukhin, V.; Lyautey, J.; Lederrey, C.; Lefort, F.; Rossier, A.; Chen, X.Q.; Anker, P. The origin and mechanism of circulating DNA. Ann. N. Y. Acad. Sci. 2000, 906, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Les acides nucléiques du plasma sanguin chez l’homme. CR Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.Z.; Magnus Hjelm, N. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar]
- Yu, S.C.Y.; Lee, S.W.Y.; Jiang, P.; Leung, T.Y.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin. Chem. 2013, 59, 1228–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Gomez-Manjon, I.; Moreno-Izquierdo, A.; Mayo, S.; Moreno-Garcia, M.; Delmiro, A.; Escribano, D.; Fernandez-Martinez, F.J. Noninvasive Prenatal Testing: Comparison of Two Mappers and Influence in the Diagnostic Yield. Biomed. Res. Int. 2018, 2018, 9498140. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol. 2014, 813, 109–122. [Google Scholar]
- Kaya, M.; Ahishali, B. Basic physiology of the blood-brain barrier in health and disease: A brief overview. Tissue Barriers 2021, 9, 1840913. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, G.; Takata, F.; Kataoka, Y.; Kanou, K.; Morichi, S.; Dohgu, S.; Kawashima, H. The neuroinflammatory role of pericytes in epilepsy. Biomedicines 2021, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Mann, A.; Ekstein, D.; Eyal, S. Breaking Bad: The Structure and Function of the Blood-Brain Barrier in Epilepsy. AAPS J. 2017, 19, 973–988. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Aronica, E.; Gorter, J.A. Blood-brain barrier dysfunction, seizures and epilepsy. Semin. Cell Dev. Biol. 2015, 38, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Erdo, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef] [Green Version]
- van Vliet, E.A.; da Costa Araújo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Lacoste, B.; Comin, C.H.; Ben-Zvi, A.; Kaeser, P.S.; Xu, X.; Costa, L.D.F.; Gu, C. Sensory-related neural activity regulates the structure of vascular networks in the cerebral cortex. Neuron 2014, 83, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Swissa, E.; Serlin, Y.; Vazana, U.; Prager, O.; Friedman, A. Blood–brain barrier dysfunction in status epileptics: Mechanisms and role in epileptogenesis. Epilepsy Behav. 2019, 101, 106285. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgard, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [Green Version]
- Mendioroz, M.; Martinez-Merino, L.; Blanco-Luquin, I.; Urdanoz, A.; Roldan, M.; Jerico, I. Liquid biopsy: A new source of candidate biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Chatterton, Z.; Mendelev, N.; Chen, S.; Carr, W.; Kamimori, G.H.; Ge, Y.; Dwork, A.J.; Haghighi, F. Bisulfite Amplicon Sequencing Can Detect Glia and Neuron Cell-Free DNA in Blood Plasma. Front. Mol. Neurosci. 2021, 14, 672614. [Google Scholar] [CrossRef] [PubMed]
- Liimatainen, S.P.; Jylhävä, J.; Raitanen, J.; Peltola, J.T.; Hurme, M.A. The concentration of cell-free DNA in focal epilepsy. Epilepsy Res. 2013, 105, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Alapirtti, T.; Jylhävä, J.; Raitanen, J.; Mäkinen, R.; Peltola, J.; Hurme, M.A.; Liimatainen, S. The concentration of cell-free DNA in video-EEG patients is dependent on the epilepsy syndrome and duration of epilepsy. Neurol. Res. 2016, 38, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Shemer, R.; Magenheim, J.; Dor, Y. Digital Droplet PCR for Monitoring Tissue-Specific Cell Death Using DNA Methylation Patterns of Circulating Cell-Free DNA. Curr. Protoc. Mol. Biol. 2019, 127, e90. [Google Scholar] [CrossRef]
- Combaret, V.; Audoynaud, C.; Iacono, I.; Favrot, M.C.; Schell, M.; Bergeron, C.; Puisieux, A. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res. 2002, 62, 3646–3648. [Google Scholar]
- Salkeni, M.A.; Zarzour, A.; Ansay, T.Y.; McPherson, C.M.; Warnick, R.E.; Rixe, O.; Bahassi, E.M. Detection of EGFRvIII mutant DNA in the peripheral blood of brain tumor patients. J. Neurooncol. 2013, 115, 27–35. [Google Scholar] [CrossRef]
- Boisselier, B.; Pérez-Larraya, J.G.; Rossetto, M.; Labussière, M.; Ciccarino, P.; Marie, Y.; Delattre, J.Y.; Sanson, M. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology 2012, 79, 1693–1698. [Google Scholar] [CrossRef]
- Ko, A.; Sim, N.S.; Choi, H.S.; Yang, D.; Kim, S.H.; Lee, J.S.; Kim, D.S.; Lee, J.H.; Kim, H.D.; Kang, H.C. Efficacy of the Ketogenic Diet for Pediatric Epilepsy According to the Presence of Detectable Somatic mTOR Pathway Mutations in the Brain. J. Clin. Neurol. 2022, 18, 71–78. [Google Scholar] [CrossRef]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-resistant epilepsy: Multiple hypotheses, few answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Bohers, E.; Viailly, P.; Jardin, F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals 2021, 14, 596. [Google Scholar] [CrossRef]
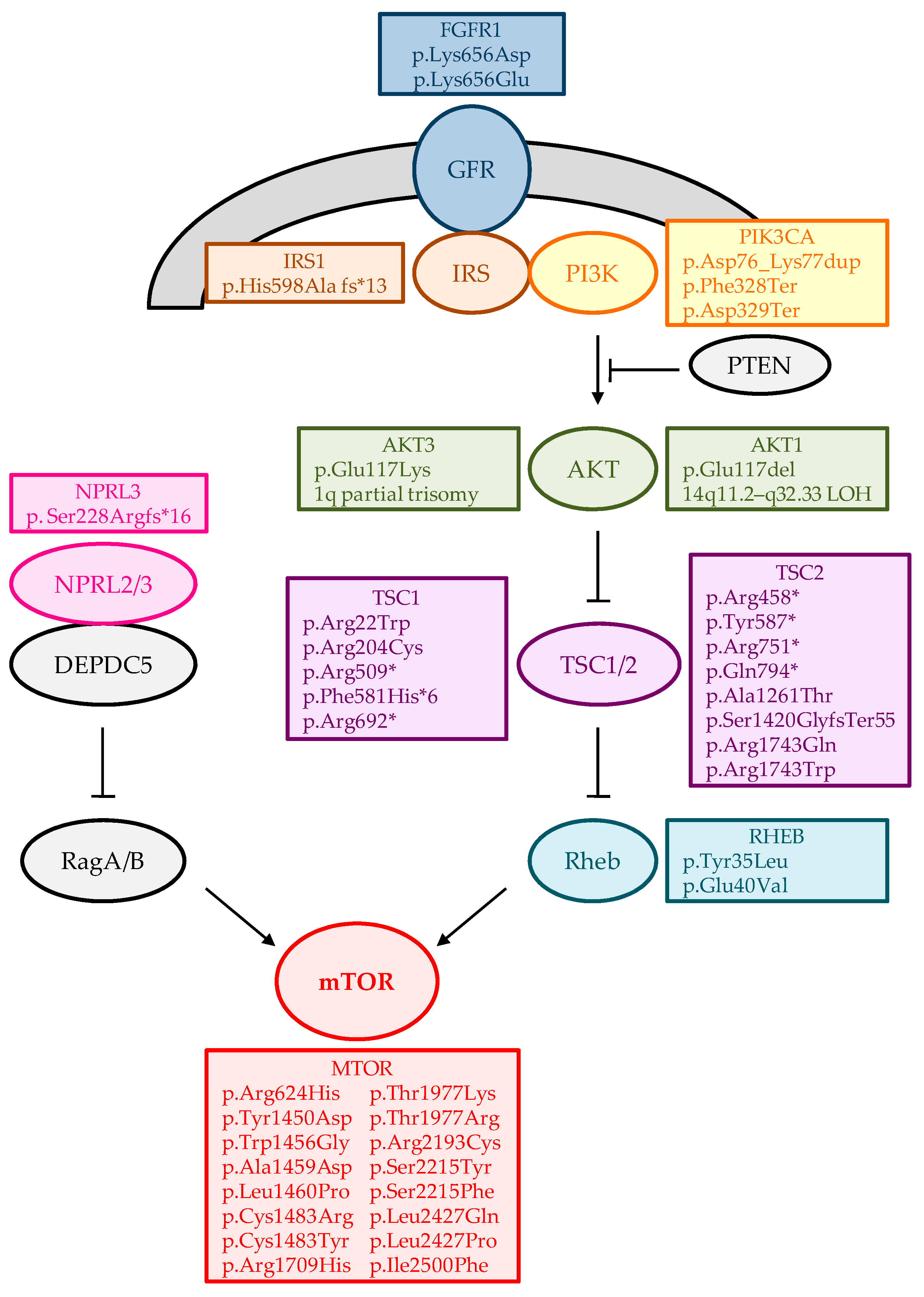
| Gene/Loci | Variant | Sample | Refractory Epilepsy | N | References |
|---|---|---|---|---|---|
| AKT1 (NM_005163) | c.349_351del; p.Glu117del | RB | FCD Iib | 1 + | [34] |
| BMP4, AKT1 | (chr14:24,419,118–106,072,470) LOH | RB | HHE | 1 | [35] |
| AKT3 | 1q21.1-q44 trisomy | RB | HME | 1 | [36] |
| AKT3 | 1q partial trisomy | RB | HME | 1 | [37] |
| AKT3 | 1q partial trisomy | RB | HME | 1 | [37] |
| AKT3 (NM_001206729) | c.49C>T; p.(Glu17Lys) | RB | HME/FCD Iia | 8 | [37,38,39,40,41,42] |
| BRAF (NM_004333) | c.1799T>A; p.(Val600Glu) | RB | GG | 14 | [33,41,43,44] |
| BRAF (NM_004333) | c.1518_1526dup | RB | GG | 1 | [43] |
| CREBBP | (chr16:0–31,543,619) LOH | RB | HHE | 2 | [35] |
| DNMT3A (NM_175629) | c.2141C>G; p.(Ser714Cys) | RB | DNT | 1 | [43] |
| FGFR1 (NM_023110) | c.1966_1968delinsGAC; p.(Lys656Asp) | RB | DNT | 1 | [43] |
| FGFR1 (NM_023110) | c.1966A>G; p.(Lys656Glu) | RB | DNT | 1 | [43] |
| GLI2, IHH, LRP2, STK36, WNT10A, WNT6 | (chr2:103,856,408–243,199,373) LOH | RB | HHE | 1 | [35] |
| GLI3(NM_000168) | c.2071C>T; p.(Gln691Ter) | RB | HHE | 1 | [35] |
| GLI3(NM_000168) | c.2989dupG; p.(Ala997GlyfsTer87) | RB | HHE | 1 | [35] |
| GLI3(NM_000168) | c.3172C>T; p.(Arg1058Ter) | RB | HHE | 1 | [35] |
| GLI3(NM_000168) | c.3442C>T; p.(Gln1148Ter) | RB | HHE | 1 | [35] |
| SHH, SMO, WNT16, WNT2 | (chr7:58,814,064–159,138,663) LOH | RB | HHE | 1 | [35] |
| GLI3, SHH, SMO, WNT16, WNT2 | (chr7:986,211–60,069,242;58,814,064–159,138,663) CNVs | RB | HHE | 1 | [35] |
| GNAQ (NM_002072) | c.548G>A; p.(Arg183Gln) | RB | ffSWS | 4 | [45] |
| HTR6 (NM_000871) | c.G469A; p.(Ala157Thr) | RB | FCD Iib | 1 | [46] |
| IRS1 (NM_005544) | c.1791dupG; p.(His598Ala fsTer13) | RB | FCD Iib | 1 * | [46] |
| KCNH1(NM_172362) | c.2138T>A; p.(Val713Glu) | RB | FCD Iib | 1 | [47] |
| KRAS (NM_004985) | c.40; G>A; p.(Val14Ile) | RB | GG and HS | 1 | [43] |
| LIS (PAFAH1B1) (NM_000430) | c.190A>T; p.(Lys64Ter) | CSF | Subcortical band heterotopia | 1 | [33] |
| MTOR (NM_004958) | c.1871G>A; p.(Arg624His) | RB | FCD Iia | 1 | [48] |
| MTOR (NM_004958) | c.4348T>G; p.(Tyr1450Asp) | RB | FCD Iib | 1 | [48] |
| MTOR (NM_004958) | c.4366T>G; p.(Trp1456Gly) | RB | FCD Iib | 2 | [41,49] |
| MTOR (NM_004958) | c.4376C>A; p.(Ala1459Asp) | RB | FCD Iia/FCD Iib | 5 | [40,50,51] |
| MTOR (NM_004958) | c.4379T>C; p.(Leu1460Pro) | RB | FCD Iia/FCD Iib | 4 | [34,40,41] |
| MTOR (NM_004958) | c.4447T>C; p.(Cys1483Arg) | RB | FCD Iib | 2 | [41,48] |
| MTOR (NM_004958) | c.4448G>A; p.(Cys1483Tyr) | RB | HME/FCD Iib | 2 | [38,41] |
| MTOR (NM_004958) | c.5126G>A; p.(Arg1709His) | RB | FCD Iia | 1 | [48] |
| MTOR (NM_004958) | c.5930C>A; p.(Thr1977Lys) | RB | FCD Iib | 8 | [40,42,43,46,48,50] |
| MTOR (NM_004958) | c.5930C>G; p.(Thr1977Arg) | RB | HME/FCD | 2 | [39] |
| MTOR (NM_004958) | c.6577C>T; p.(Arg2193Cys) | RB | FCD Iia | 1 | [48] |
| MTOR (NM_004958) | c.6644C>A; p.(Ser2215Tyr) | RB | FCD Iia/FCD Iib | 9 | [40,41,42,50] |
| MTOR (NM_004958) | c.6644C>T; p.(Ser2215Phe) | RB | HME/FCD Iia/FCD Iib/Polymicrogyria/SKS | 16 | [39,40,41,48,52] |
| MTOR (NM_004958) | c.7280T>A; p.(Leu2427Gln) | RB | FCD Iia/FCD Iib | 4 | [41,48] |
| MTOR (NM_004958) | c.7280T>C; p.(Leu2427Pro) | RB | FCD Iia | 2 | [41] |
| MTOR (NM_004958) | c.7498A>T; p.(Ile2500Phe) | RB | FCD Iia | 1 | [40] |
| NF1 (NM_000267) | c.2674del; p.(Ser892AlafsTer10) | RB | HS | 1 | [43] |
| NPRL3 (NM_001077350) | c.682_683dup; p.(Ser228ArgfsTer16) | RB | FCD Iia | 1 | [43] |
| PIK3CA(NM_006218) | c.1624G>A; p.(Glu542Lys) | RB | HME/FCD Iia | 3 | [39,40] |
| PIK3CA(NM_006218) | c.1633G>A; p.(Glu545Lys) | RB/CSF | HME | 6 | [38,41,44] |
| PIK3CA(NM_006218) | c.3140A>G; p.(His1047Arg) | RB | HME/FCD Iia | 2 | [40] |
| PRKACA(NM_002730) | c.226-231dup; p.(Asp76_Lys77dup) | RB | HHE | 1 | [35] |
| PRKACA(NM_002730) | c.983_984delTT; p.(Phe328Ter) | RB | HHE | 1 | [35] |
| PRKACA(NM_002730) | c.984dupT; p.(Asp329Ter) | RB | HHE | 1 | [35] |
| RAB6B (NM_016577) | c.C383T; p.(Thr128Met) | RB | FCD Iia | 1 | [46] |
| RALA (NM_005402) | c.G482A; p.(Arg161Gln) | RB | FCD Iib | 1 | [46] |
| RHEB (NM_005614) | c.[105C>A,104A>T]; p.(Tyr35Leu) | RB | HME/FCD Iib | 1 | [40] |
| RHEB (NM_005614) | c.119A>T; p.(Glu40Val) | RB | HME/FCD Iib | 2 | [40,53] |
| SLC35A2 (NM_005660) | c.935C>T; p.(Ser312Phe) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.112_116delinsTGGTGGTCCAGAATG; p.(Ile38TrpfsTer59) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.206C>T; p.(Thr69Ile) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.275-1G>T | RB | LGS/MOGHE | 1 | [54,55] |
| SLC35A2 (NM_005660) | c.335_339dupCGCTC; p.(Lys114ArgfsTer32) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.359_360delTC; p.(Leu120HisfsTer7) | RB | MOGHE | 2 | [54] |
| SLC35A2 (NM_005660) | c.359T>C; p.(Leu120Pro) | RB | MOGHE | 1 | [41,54] |
| SLC35A2 (NM_005660) | c.385C>T; p.(Gln129Ter) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.502G>A; p.(Gln168Ter) | RB/CSF | LGS/MOGHE | 1 | [44,54,55] |
| SLC35A2 (NM_005660) | c.553C>T; p.(Gln185Ter) | RB | LGS/MOGHE | 2 | [54,55] |
| SLC35A2 (NM_005660) | c.569_572delGAGG; p.(Gly190AlafsTer158) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.580_616dupCCACTGGATCAGAACCCTGGGGCAGGCCTGGCAGCCG; p.(Val206AlafsTer28) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.589C>T; p.(Gln197Ter) | RB | LGS/MOGHE | 1 | [54,55] |
| SLC35A2 (NM_005660) | c.603_606dupAGGC; p.(Leu203ArgfsTer20) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.634_635delTC; p.(Ser212LeufsTer9) | RB | mMCD/MOGHE/NLFE/WS | 3 | [40,54,56,57] |
| SLC35A2 (NM_005660) | c.671T>C; p.(Leu224Pro) | RB | MOGHE | 1 | [41,54] |
| SLC35A2 (NM_005660) | c.703A>C; p.(Asn235His) | RB | LGS/MOGHE | 1 | [54,55] |
| SLC35A2 (NM_005660) | c.760G>T; p.(Glu254Ter) | RB | LGS/MOGHE | 1 | [54,55] |
| SLC35A2 (NM_005660) | c.801C>G; p.(Tyr267Ter) | RB | mMCD/MOGHE | 1 | [40,54] |
| SLC35A2 (NM_005660) | c.804dupA; p.(Pro269ThrfsTer24) | RB | mMCD/MOGHE | 1 | [40,54] |
| SLC35A2 (NM_005660) | c.842G>A; p.(Gly281Asp) | RB | MOGHE | 1 | [41,54] |
| SLC35A2 (NM_005660) | c.886_888delCTC; p.(Leu296del) | RB | mMCD/MOGHE | 1 | [40,54] |
| SLC35A2 (NM_005660) | c.905C>T; p.(Ser302Phe) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | c.918_929delGCTGTCCACTGT; p.(Leu307_Val310del) | RB | MOGHE | 1 | [54] |
| SLC35A2 (NM_005660) | p.(Cys210Tyr) | RB | MOGHE | 1 | [42] |
| SLC35A2 (NM_005660) | p.(Pro15Thr) | RB | MOGHE | 1 | [42] |
| SLC35A2 (NM_005660) | c.164G>T; p.(Arg55Leu) | RB | MCD | 1 | [56] |
| SLC35A2 (NM_005660) | c.339_340insCTC; p.(Leu113dup) | RB | NLFE | 1 | [56] |
| SLC35A2 (NM_005660) | c.747_757dup; p.(Ala253GlyfsTer100) | RB | MCD | 1 | [56] |
| SLC35A2 (NM_005660) | c.910T>C; p.(Ser304Pro) | RB | NLFE | 1 | [56] |
| TSC1 (NM_000368) | c.1525C>T; p.(Arg509Ter) | RB | FCD Iib | 1 | [41] |
| TSC1 (NM_000368) | c.2074C>T; p.(Arg692Ter) | RB | FCD Iib | 1 | [41] |
| TSC1 (NM_000368) | c.610C>T; p.(Arg204Cys) | RB | FCD Iia | 1 | [41] |
| TSC1 (NM_000368) | c.64C>T; p.(Arg22Trp) | RB | FCD Iib | 1 | [58] |
| TSC1 (NM_000368) | c.1741_1742delTT; p.(Phe581HisTer6) | CSF | FCD Iib | 1 | [33] |
| TSC2 (NM_000548) | c.1372C>T; p.(Arg458Ter) | RB | FCD Iib | 1 | [41] |
| TSC2 (NM_000548) | c.1754_1755delGT; p.(Tyr587Ter) | RB | HME | 1 | [39] |
| TSC2 (NM_000548) | c.2251C>T; p.(Arg751Ter) | RB | FCD | 1 | [39] |
| TSC2 (NM_000548) | c.2380C>T; p.(Gln794Ter) | RB | FCD Iib | 1 | [40] |
| TSC2 (NM_000548) | c.4258_4261delCAGT; p.(Ser1420GlyfsTer55) | RB | FCD Iib | 1 | [58] |
| TSC2 (NM_000548) | c.5228G>A; p.(Arg1743Gln) | RB | FCD Iib | 1 | [40] |
| TSC2 (NM_000548) | c.3781G>A; p.(Ala1261Thr) | RB | FCD Iib | 1 + | [34] |
| TSC2 (NM_000548) | c.5227C>T; p.(Arg1743Trp) | RB | FCD Iib | 1 | [46] |
| WNT11 | (chr11:64879188–135006516) LOH | RB | HHE | 1 | [35] |
| ZNF337 (NM_001290261) | c.692_693del; p.(Thr231Arg fsTer45) | RB | FCD Iib | 1 * | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayo, S.; Gómez-Manjón, I.; Fernández-Martínez, F.J.; Camacho, A.; Martínez, F.; Benito-León, J. CfDNA Measurement as a Diagnostic Tool for the Detection of Brain Somatic Mutations in Refractory Epilepsy. Int. J. Mol. Sci. 2022, 23, 4879. https://doi.org/10.3390/ijms23094879
Mayo S, Gómez-Manjón I, Fernández-Martínez FJ, Camacho A, Martínez F, Benito-León J. CfDNA Measurement as a Diagnostic Tool for the Detection of Brain Somatic Mutations in Refractory Epilepsy. International Journal of Molecular Sciences. 2022; 23(9):4879. https://doi.org/10.3390/ijms23094879
Chicago/Turabian StyleMayo, Sonia, Irene Gómez-Manjón, Francisco Javier Fernández-Martínez, Ana Camacho, Francisco Martínez, and Julián Benito-León. 2022. "CfDNA Measurement as a Diagnostic Tool for the Detection of Brain Somatic Mutations in Refractory Epilepsy" International Journal of Molecular Sciences 23, no. 9: 4879. https://doi.org/10.3390/ijms23094879
APA StyleMayo, S., Gómez-Manjón, I., Fernández-Martínez, F. J., Camacho, A., Martínez, F., & Benito-León, J. (2022). CfDNA Measurement as a Diagnostic Tool for the Detection of Brain Somatic Mutations in Refractory Epilepsy. International Journal of Molecular Sciences, 23(9), 4879. https://doi.org/10.3390/ijms23094879