
The Role of Epigenetics in Primary Biliary Cholangitis

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
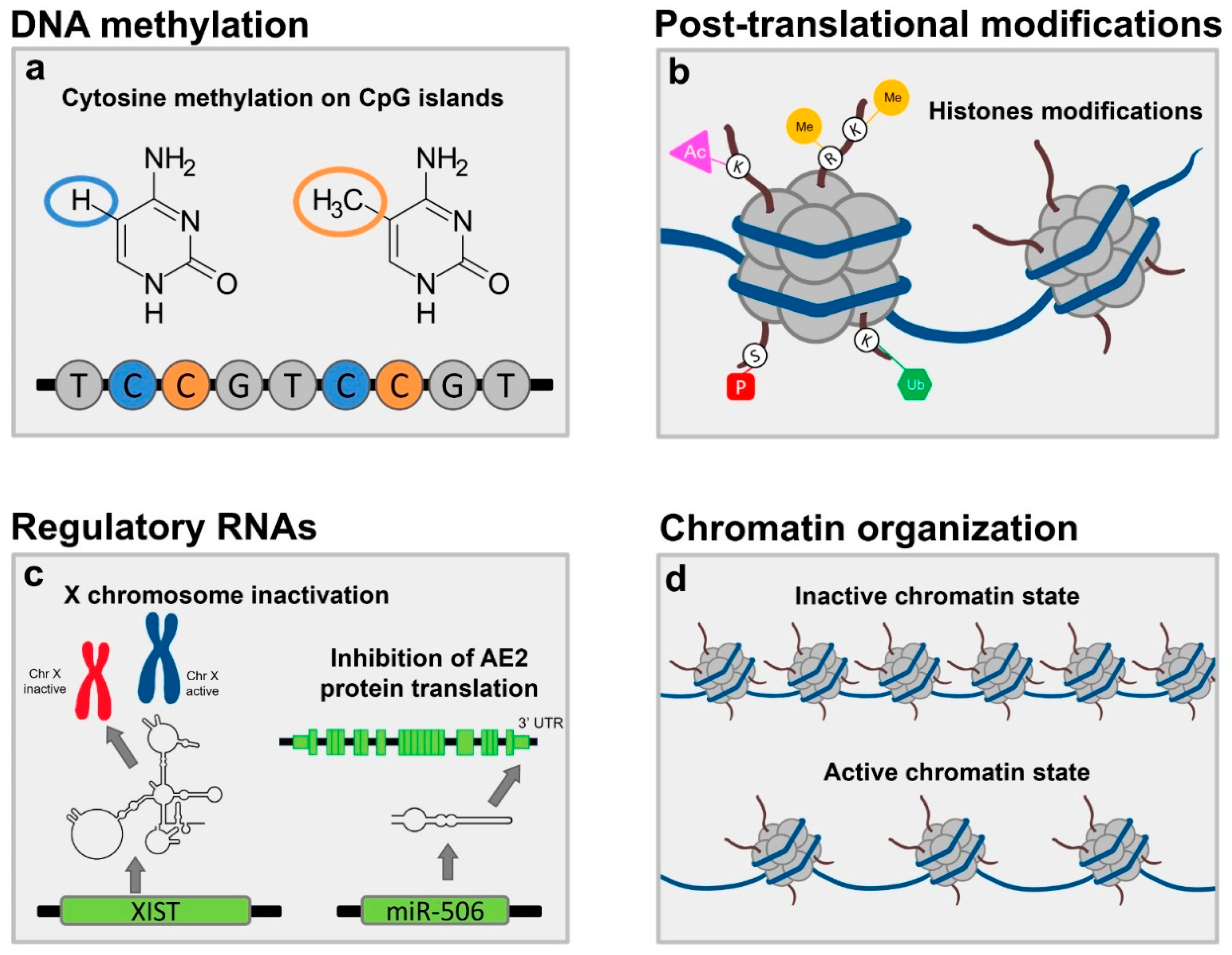
2. Traditional Pillars of Epigenetics
2.1. DNA Methylation
2.2. Post Translational Modifications to Histone Proteins
2.3. Non-Coding RNA Expression
3. ChrX Monosomy and Inactivation
4. XWAS Results and How They Associate with Epigenetics
5. The Role of the Environment
6. Future Perspectives and Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A
- (1)
- GENCODE [90]; we selected the comprehensive gene annotation of lncRNA genes on chromosome X, version 25 (https://www.gencodegenes.org/releases/current.html accessed on 1 December 2018);
- (2)
- miRbase [91,92,93,94]; we selected the gene annotation for miRNAs version 20. (http://www.mirbase.org/, accessed on 1 December 2018);
- (3)
References
- Gerussi, A.; Cristoferi, L.; Carbone, M.; Asselta, R.; Invernizzi, P. The immunobiology of female predominance in primary biliary cholangitis. J. Autoimmun. 2018, 95, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Orphanet: An Online Database of Rare Diseases and Orphan Drugs. Copyright, INSERM 1997. Available online: http://www.orpha.net (accessed on 27 February 2022).
- Smyk, D.S.; Rigopoulou, E.I.; Pares, A.; Billinis, C.; Burroughs, A.K.; Muratori, L.; Invernizzi, P.; Bogdanos, D.P. Sex Differences Associated with Primary Biliary Cirrhosis. Clin. Dev. Immunol. 2012, 2012, e610504. [Google Scholar] [CrossRef]
- Carbone, M.; Mells, G.F.; Pells, G.; Dawwas, M.F.; Newton, J.L.; Heneghan, M.A.; Neuberger, J.M.; Day, D.B.; Ducker, S.J.; Sandford, R.N.; et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology 2013, 144, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Wang, G.-Q.; Gershwin, M.E.; Hirschfield, G.M. Primary biliary cholangitis. Lancet 2020, 396, 1915–1926. [Google Scholar] [CrossRef]
- Galoosian, A.; Hanlon, C.; Zhang, J.; Holt, E.W.; Yimam, K.K. Clinical Updates in Primary Biliary Cholangitis: Trends, Epidemiology, Diagnostics, and New Therapeutic Approaches. J. Clin. Transl. Hepatol. 2020, 8, 49–60. [Google Scholar] [CrossRef]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Mackay, I.R.; Gores, G.J.; Ansari, A.A.; Van de Water, J.; Gershwin, E. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009, 49, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, S.; Miyakawa, H.; Nakamura, M.; Ishibashi, H.; Kikuchi, K.; Kita, H.; Niiro, H.; Arinobu, Y.; Ono, N.; Mackay, I.R.; et al. CD4 T-cell autoreactivity to the mitochondrial autoantigen PDC-E2 in AMA-negative primary biliary cirrhosis. J. Autoimmun. 2008, 31, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Oukka, M.; Kuchroo, V.K. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007, 8, 345–350. [Google Scholar] [CrossRef]
- Ueno, H. T follicular helper cells in human autoimmunity. Curr. Opin. Immunol. 2016, 43, 24–31. [Google Scholar] [CrossRef]
- Gerussi, A.; Carbone, M.; Corpechot, C.; Schramm, C. The genetic architecture of primary biliary cholangitis. Eur. J. Med. Genet. 2021, 64, 104292. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Chrétien, Y.; Chazouillères, O.; Poupon, R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J. Hepatol. 2010, 53, 162–169. [Google Scholar] [CrossRef]
- Marzorati, S.; Lleo, A.; Carbone, M.; Gershwin, M.E.; Invernizzi, P. The epigenetics of PBC: The link between genetic susceptibility and environment. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 650–659. [Google Scholar] [CrossRef]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.; Zhao, K. The epigenetic basis of cellular heterogeneity. Nat. Rev. Genet. 2021, 22, 235–250. [Google Scholar] [CrossRef]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Liao, J.; Invernizzi, P.; Zhao, M.; Bernuzzi, F.; Ma, L.; Lanzi, G.; Ansari, A.A.; Coppel, R.L.; Zhang, P.; et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology 2012, 55, 153–160. [Google Scholar] [CrossRef]
- Alabraba, E.B.; Lai, V.; Boon, L.; Wigmore, S.J.; Adams, D.H.; Afford, S.C. Coculture of human liver macrophages and cholangiocytes leads to CD40-dependent apoptosis and cytokine secretion. Hepatology 2008, 47, 552–562. [Google Scholar] [CrossRef]
- Carbone, M.; Milani, C.; Gerussi, A.; Ronca, V.; Cristoferi, L.; Invernizzi, P. Primary biliary cholangitis: A multifaceted pathogenesis with potential therapeutic targets. J. Hepatol. 2020, 73, 965–966. [Google Scholar] [CrossRef]
- Gerussi, A.; Carbone, M. Primary Biliary Cholangitis. Autoimmune Liver Dis. 2020, 123–141. [Google Scholar]
- Asselta, R.; Paraboschi, E.M.; Gerussi, A.; Cordell, H.J.; Mells, G.F.; Sandford, R.N.; Jones, D.E.; Nakamura, M.; Ueno, K.; Hitomi, Y.; et al. X Chromosome Contribution to the Genetic Architecture of Primary Biliary Cholangitis. Gastroenterology 2021, 160, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Cavaciocchi, F.; Lleo, A.; Cheroni, C.; De Francesco, R.; Lombardi, S.A.; De Santis, M.; Meda, F.; Raimondo, M.G.; Crotti, C.; et al. Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Front. Immunol. 2014, 5, 128. [Google Scholar] [CrossRef]
- Lleo, A.; Zhang, W.; Zhao, M.; Tan, Y.; Bernuzzi, F.; Zhu, B.; Liu, Q.; Tan, Q.; Malinverno, F.; Valenti, L.; et al. DNA methylation profiling of the X chromosome reveals an aberrant demethylation on CXCR3 promoter in primary biliary cirrhosis. Clin. Epigenetics 2015, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-H.; Lian, Z.-X.; Cheng, C.-M.; Lan, R.Y.; Yang, G.-X.; Moritoki, Y.; Chiang, B.-L.; Ansari, A.A.; Tsuneyama, K.; Coppel, R.L.; et al. Increased levels of chemokine receptor CXCR3 and chemokines IP-10 and MIG in patients with primary biliary cirrhosis and their first degree relatives. J. Autoimmun. 2005, 25, 126–132. [Google Scholar] [CrossRef]
- Karin, N. CXCR3 Ligands in Cancer and Autoimmunity, Chemoattraction of Effector T Cells, and Beyond. Front. Immunol. 2020, 11, 976. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Huang, Y.; Liu, Y.; Sun, Y.; Zhou, Y.; Gu, M.; Chen, Y.; Xia, R.; Chen, S.; Deng, A.; et al. β-arrestin 1 modulates functions of autoimmune T cells from primary biliary cirrhosis patients. J. Clin. Immunol. 2011, 31, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.-H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2014, 518, 337. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Perugorria, M.J.; Santos-Laso, A.; Bujanda, L.; Beuers, U.; Banales, J.M. Primary biliary cholangitis: A tale of epigenetically-induced secretory failure? J. Hepatol. 2018, 69, 1371–1383. [Google Scholar] [CrossRef]
- Salas, J.T.; Banales, J.M.; Sarvide, S.; Recalde, S.; Ferrer, A.; Uriarte, I.; Oude Elferink, R.P.; Prieto, J.; Medina, J.F. Ae2a, b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology 2008, 134, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.F.; Martínez-Ansó; Vazquez, J.J.; Prieto, J. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 1997, 25, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Sáez, E.; Úriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; et al. Up-regulation of microRNA 506 leads to decreased Cl−/HCO3− anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Erice, O.; Munoz-Garrido, P.; Vaquero, J.; Perugorria, M.J.; Fernandez-Barrena, M.G.; Saez, E.; Santos-Laso, A.; Arbelaiz, A.; Jimenez-Agüero, R.; Fernandez-Irigoyen, J.; et al. MicroRNA-506 promotes primary biliary cholangitis–like features in cholangiocytes and immune activation. Hepatology 2018, 67, 1420–1440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Barbier, O.; Smalling, R.; Tsuchiya, H.; Lee, S.; Delker, D.; Zou, A.; Hagedorn, C.H.; Wang, L. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci. Rep. 2016, 6, 20559. [Google Scholar] [CrossRef]
- Wang, Y.; Hylemon, P.B.; Zhou, H. Long Noncoding RNA H19: A Key Player in Liver Diseases. Hepatology 2021, 74, 1652–1659. [Google Scholar] [CrossRef]
- Pang, K.C.; Dinger, M.E.; Mercer, T.R.; Malquori, L.; Grimmond, S.M.; Chen, W.; Mattick, J.S. Genome-wide identification of long noncoding RNAs in CD8+ T cells. J. Immunol. 2009, 182, 7738–7748. [Google Scholar] [CrossRef]
- Padgett, K.A.; Lan, R.Y.; Leung, P.C.; Lleo, A.; Dawson, K.; Pfeiff, J.; Mao, T.K.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Primary biliary cirrhosis is associated with altered hepatic microRNA expression. J. Autoimmun. 2009, 32, 246–253. [Google Scholar] [CrossRef]
- Qin, B.; Huang, F.; Liang, Y.; Yang, Z.; Zhong, R. Analysis of altered microRNA expression profiles in peripheral blood mononuclear cells from patients with primary biliary cirrhosis. J. Gastroenterol. Hepatol. 2013, 28, 543–550. [Google Scholar] [CrossRef]
- Ninomiya, M.; Kondo, Y.; Funayama, R.; Nagashima, T.; Kogure, T.; Kakazu, E.; Kimura, O.; Ueno, Y.; Nakayama, K.; Shimosegawa, T. Distinct microRNAs expression profile in primary biliary cirrhosis and evaluation of miR 505-3p and miR197-3p as novel biomarkers. PLoS ONE 2013, 8, e66086. [Google Scholar] [CrossRef]
- Tan, Y.; Pan, T.; Ye, Y.; Ge, G.; Chen, L.; Wen, D.; Zou, S. Serum MicroRNAs as Potential Biomarkers of Primary Biliary Cirrhosis. PLoS ONE 2014, 9, e111424. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Morishita, A.; Nomura, T.; Tani, J.; Miyoshi, H.; Yoneyama, H.; Iwama, H.; Himoto, T.; Masaki, T. Identification of microRNA profiles associated with refractory primary biliary cirrhosis. Mol. Med. Rep. 2016, 14, 3350–3356. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-Y.; Hou, Y.-Q.; Luo, L.-J.; Ao, L. Altered expression of miR-92a correlates with Th17 cell frequency in patients with primary biliary cirrhosis. Int. J. Mol. Med. 2016, 38, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Strachan, T. Genetics and Genomics in Medicine; Garland Science: New York, NY, USA, 2015; ISBN 9780815344803. [Google Scholar]
- Dossin, F.; Pinheiro, I.; Żylicz, J.J.; Roensch, J.; Collombet, S.; Le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Bondy, C.A.; Cheng, C. Monosomy for the X chromosome. Chromosom. Res. 2009, 17, 649–658. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Tukiainen, T.; Villani, A.-C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Du, S.; Itoh, N.; Askarinam, S.; Hill, H.; Arnold, A.P.; Voskuhl, R.R. XY sex chromosome complement, compared with XX, in the CNS confers greater neurodegeneration during experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2014, 111, 2806–2811. [Google Scholar] [CrossRef]
- Golden, L.C.; Itoh, Y.; Itoh, N.; Iyengar, S.; Coit, P.; Salama, Y.; Arnold, A.P.; Sawalha, A.H.; Voskuhl, R.R. Parent-of-origin differences in DNA methylation of X chromosome genes in T lymphocytes. Proc. Natl. Acad. Sci. USA 2019, 116, 26779–26787. [Google Scholar] [CrossRef] [PubMed]
- Jowhar, Z.; Shachar, S.; Gudla, P.R.; Wangsa, D.; Torres, E.; Russ, J.L.; Pegoraro, G.; Ried, T.; Raznahan, A.; Misteli, T. Effects of human sex chromosome dosage on spatial chromosome organization. Mol. Biol. Cell 2018, 29, 2458–2469. [Google Scholar] [CrossRef] [PubMed]
- Di, K.N.; Disteche, C.M. Dosage compensation of the active X chromosome in mammals. Nat. Genet. 2006, 38, 47–53. [Google Scholar] [CrossRef]
- Gupta, V.; Parisi, M.; Sturgill, D.; Nuttall, R.; Doctolero, M.; Dudko, O.K.; Malley, J.D.; Eastman, P.S.; Oliver, B. Global analysis of X-chromosome dosage compensation. J. Biol. 2006, 5, 3. [Google Scholar] [CrossRef]
- Oliva, M.; Muñoz-Aguirre, M.; Kim-Hellmuth, S.; Wucher, V.; Gewirtz, A.D.H.; Cotter, D.J.; Parsana, P.; Kasela, S.; Balliu, B.; Viñuela, A.; et al. The impact of sex on gene expression across human tissues. Science 2020, 369, eaba3066. [Google Scholar] [CrossRef]
- Kirsch-Volders, M.; Bolognesi, C.; Ceppi, M.; Bruzzone, M.; Fenech, M. Micronuclei, inflammation and auto-immune disease. Mutat. Res. Mutat. Res. 2020, 786, 108335. [Google Scholar] [CrossRef]
- Svyryd, Y.; Hernández-Molina, G.; Vargas, F.; Sánchez-Guerrero, J.; Segovia, D.A.; Mutchinick, O.M. X chromosome monosomy in primary and overlapping autoimmune diseases. Autoimmun. Rev. 2012, 11, 301–304. [Google Scholar] [CrossRef]
- Leach, N.T.; Rehder, C.; Jensen, K.; Holt, S.; Jackson-Cook, C. Human chromosomes with shorter telomeres and large heterochromatin regions have a higher frequency of acquired somatic cell aneuploidy. Mech. Ageing Dev. 2004, 125, 563–573. [Google Scholar] [CrossRef]
- Invernizzi, P.; Miozzo, M.; Battezzati, P.M.; Bianchi, I.; Grati, F.R.; Simoni, G.; Selmi, C.; Watnik, M.; Gershwin, M.E.; Podda, M. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004, 363, 533–535. [Google Scholar] [CrossRef]
- Miozzo, M.; Selmi, C.; Gentilin, B.; Grati, F.R.; Sirchia, S.; Oertelt, S.; Zuin, M.; Gershwin, M.E.; Podda, M.; Invernizzi, P. Preferential X chromosome loss but random inactivation characterize primary biliary cirrhosis. Hepatology 2007, 46, 456–462. [Google Scholar] [CrossRef]
- Ford, J.H.; Schultz, C.J.; Correll, A.T. Chromosome elimination in micronuclei: A common cause of hypoploidy. Am. J. Hum. Genet. 1988, 43, 733–740. [Google Scholar]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Catalán, J.; Autio, K.; Kuosma, E.; Norppa, H. Age-dependent inclusion of sex chromosomes in lymphocyte micronuclei of man. Am. J. Hum. Genet. 1998, 63, 1464–1472. [Google Scholar] [CrossRef]
- Guttenbach, M.; Schakowski, R.; Schmid, M. Aneuploidy and ageing: Sex chromosome exclusion into micronuclei. Hum. Genet. 1994, 94, 295–298. [Google Scholar] [CrossRef]
- Gao, F.; Chang, D.; Biddanda, A.; Ma, L.; Guo, Y.; Zhou, Z.; Keinan, A. XWAS: A Software Toolset for Genetic Data Analysis and Association Studies of the X Chromosome. J. Hered. 2015, 106, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, I.; Lleo, A.; Gershwin, M.E.; Invernizzi, P. The X chromosome and immune associated genes. J. Autoimmun. 2012, 38, J187–J192. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Lin, C. Enhancer, epigenetics, and human disease. Curr. Opin. Genet. Dev. 2016, 36, 27–33. [Google Scholar] [CrossRef]
- Yamagata, K.; Nakayamada, S.; Tanaka, Y. Critical roles of super-enhancers in the pathogenesis of autoimmune diseases. Inflamm. Regen. 2020, 40, 16. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.M.; Lleo, A.; Zammataro, L.; Mayo, M.J.; Invernizzi, P.; Bach, N.; Shimoda, S.; Gordon, S.C.; Podda, M.; Eric Gershwin, M.; et al. Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics 2011, 6, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Li, Z.; Wang, T.; Zhao, Y.; Wang, Y. Microarray Expression Profile of Circular RNAs in Plasma from Primary Biliary Cholangitis Patients. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 1271–1281. [Google Scholar] [CrossRef]
- Burroughs, A.K.; Rosenstein, I.J.; Epstein, O.; Hamilton-Miller, J.M.T.; Brumfitt, W.; Sherlock, S. Bacteriuria and primary biliary cirrhosis. Gut 1984, 25, 133–137. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Selmi, C.; Worman, H.J.; Gold, E.B.; Watnik, M.; Utts, J.; Lindor, K.D.; Kaplan, M.M.; Vierling, J.M. Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients. Hepatology 2005, 42, 1194–1202. [Google Scholar] [CrossRef]
- Shimoda, S.; Nakamura, M.; Ishibashi, H.; Hayashida, K.; Niho, Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: Evidence of molecular mimicry in human autoimmune diseases. J. Exp. Med. 1995, 181, 1835–1845. [Google Scholar] [CrossRef]
- Selmi, C.; Balkwill, D.L.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Kenny, T.P.; Van De Water, J.; Nantz, M.H.; et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology 2003, 38, 1250–1257. [Google Scholar] [CrossRef]
- Xu, L.; Shen, Z.; Guo, L.; Fodera, B.; Keogh, A.; Joplin, R.; O’Donnell, B.; Aitken, J.; Carman, W.; Neuberger, J.; et al. Does a betaretrovirus infection trigger primary biliary cirrhosis? Proc. Natl. Acad. Sci. USA 2003, 100, 8454–8459. [Google Scholar] [CrossRef]
- Prince, M.I.; Chetwynd, A.; Diggle, P.; Jarner, M.; Metcalf, J.V.; James, O.F. The geographical distribution of primary biliary cirrhosis in a well-defined cohort. Hepatology 2001, 34, 1083–1088. [Google Scholar] [CrossRef]
- McNally, R.J.Q.; James, P.W.; Ducker, S.; Norman, P.D.; James, O.F.W. No rise in incidence but geographical heterogeneity in the occurrence of primary biliary cirrhosis in North East England. Am. J. Epidemiol. 2014, 179, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Stanca, C.M.; Bu-Ghanim, M.; Ahmado, I.; Branch, A.D.; Schiano, T.D.; Odin, J.A.; Bach, N. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology 2006, 43, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Blain, A.; Foster Shirley, M.D.; Hudson, M.; Rushton, S.; Jeffreys Jones, D.E. Geo-epidemiology and environmental co-variate mapping of primary biliary cholangitis and primary sclerosing cholangitis. JHEP Rep. 2021, 3, 100202. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Leung, P.S.; Gershwin, M.E. Environmental basis of primary biliary cholangitis. Exp. Biol. Med. 2018, 243, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Probert, P.M.; Leitch, A.C.; Dunn, M.P.; Meyer, S.K.; Palmer, J.M.; Abdelghany, T.M.; Lakey, A.F.; Cooke, M.P.; Talbot, H.; Wills, C.; et al. Identification of a xenobiotic as a potential environmental trigger in primary biliary cholangitis. J. Hepatol. 2018, 69, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Griffiths-Jones, S. The microRNA Registry. Nucleic Acids Res. 2004, 32, D109–D111. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef]


{kind=link}
| miRNAs | Tissue | Notes | Reference |
|---|---|---|---|
| miR-122a, miR-26a, miR-328, miR-299-5p | Liver | miR-122a/miR-26 DOWN miR-328/miR-299-5p UP | Padgett et al., 2009 [39] |
| miR-506 | Liver | miR-506 UP | Banales et al., 2012 [34] |
| miR-15a-5p, miR-20a-5p, miR-140-3p, miR-106b-5p, miR-3654, miR-181a-5p | PBMCs | abnormal expression of 17 miRNAs that control cell differentiation and signal transduction | Qin et al., 2013 [40] |
| hsa-miR-505-5p, hsa-miR-141-3p, has-miR-26b-5p | Sera | hsa-miR-505-3p/miR-197-3p DOWN | Ninomiya et al., 2013 [41] |
| hsa-miR-122-5p, hsa-miR-141-3p, hsa-miR-26b-5p | Sera | disease biomarkers | Tan et al., 2014 [42] |
| miRNA-122, miRNA-378, miRNA-4311, miRNA-4714-3p | Sera | risk stratification | Sakamoto et al., 2016 [43] |
| miR-92a | Sera, PBMC | pathogenesis (Th17 cell differentiation) | Liang et al., 2016 [44] |
| Gene | Study Design | Analyzed Samples | Methylation Status | Expression in Liver/Blood (TPM) |
|---|---|---|---|---|
| PIN4 | MZ twins | serum | Variable | 5.89/0.94 |
| NHS | MZ twins | serum | Hyper-methylated | 0.12/0.25 |
| IL1RAPL2 | cases vs. controls | serum (CD14+) | Hyper-methylated | 0.17/0.10 |
| SHROOM2 | cases vs. controls | serum (CD4+) | Hypo-methylated | 1.89/0.030 |
| ATP6AP2 | cases vs. controls | serum (CD4+) | Hypo-methylated | 33.37/55.74 |
| PQBP1 | cases vs. controls | serum (CD4+) | Hyper-methylated | 35.06/31.37 |
| MAGEB2 | cases vs. controls | serum (CD8+) | Hypo-methylated | Not expressed |
| NSDHL | cases vs. controls | serum (CD14+) | Hypo-methylated | 20.97/5.21 |
| PIM2 | cases vs. controls | serum (CD14+) | Hyper-methylated | 4.80/60.37 |
| Set | Regions (n) | Regions § (kb) | DNA Content (Mb) | LncRNAs (n) | CircRNAs (n) | MiRNAs (n) | SEs (n) |
|---|---|---|---|---|---|---|---|
| PBC set | 62 | 150.3 | 9.3 | 22 | 362 | 9 | 351 |
| SD | - | 116.9 | - | - | - | - | - |
| Random sets * | 62.0 | 152.8 | 9.3 | 18.6 | 177.0 | 10.0 | 370.1 |
| SD | - | - | - | 4.6 | 78.0 | 7.2 | 51.4 |
| % ** | - | - | - | 24.5 | 5.1 | 46.5 | 63.6 |
| p value | - | - | - | 0.25 | 0.05 | 0.47 | 0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerussi, A.; Paraboschi, E.M.; Cappadona, C.; Caime, C.; Binatti, E.; Cristoferi, L.; Asselta, R.; Invernizzi, P. The Role of Epigenetics in Primary Biliary Cholangitis. Int. J. Mol. Sci. 2022, 23, 4873. https://doi.org/10.3390/ijms23094873
Gerussi A, Paraboschi EM, Cappadona C, Caime C, Binatti E, Cristoferi L, Asselta R, Invernizzi P. The Role of Epigenetics in Primary Biliary Cholangitis. International Journal of Molecular Sciences. 2022; 23(9):4873. https://doi.org/10.3390/ijms23094873
Chicago/Turabian StyleGerussi, Alessio, Elvezia Maria Paraboschi, Claudio Cappadona, Chiara Caime, Eleonora Binatti, Laura Cristoferi, Rosanna Asselta, and Pietro Invernizzi. 2022. "The Role of Epigenetics in Primary Biliary Cholangitis" International Journal of Molecular Sciences 23, no. 9: 4873. https://doi.org/10.3390/ijms23094873
APA StyleGerussi, A., Paraboschi, E. M., Cappadona, C., Caime, C., Binatti, E., Cristoferi, L., Asselta, R., & Invernizzi, P. (2022). The Role of Epigenetics in Primary Biliary Cholangitis. International Journal of Molecular Sciences, 23(9), 4873. https://doi.org/10.3390/ijms23094873