
Advanced Gene-Targeting Therapies for Motor Neuron Diseases and Muscular Dystrophies



Abstract
:1. Introduction
1.1. Gene Replacement Therapy (GRT)
1.2. Antisense Oligonucleotides (ASOs)
1.3. RNA Interference (RNAi)
1.4. CRISPR-Associated (Cas) Systems
2. Amyotrophic Lateral Sclerosis (ALS)
2.1. Clinical Manifestations and Genetic Background
2.2. Available Gene Therapies for ALS
2.2.1. GRT in ALS
2.2.2. ASOs in ALS
An ASO RNase H1-Mediated Inhibitor of SOD1: Tofersen
An ASO RNase H1-Mediated Inhibitor of ATXN2 mRNA: BIIB105
An ASO RNase H1-Mediated Inhibitor of C9ORF72 RNA: BIIB078
A Stereopure ASO RNase H1-Mediated Inhibitor of C9ORF72 RNA: WVE-004
A Patient-Specific ASO Targeting the FUS Mutation p.P525 L: Jacifusen
2.2.3. RNAi in ALS
A miRNA Inhibitor of SOD1 mRNA: SOD1-rAAVRh10.mi-SOD1
3. Spinal Muscular Atrophy (SMA)
3.1. Clinical Manifestations and Genetic Background
3.2. Available Gene Therapies for SMA
3.2.1. GRT in SMA
3.2.2. ASOs in SMA
A Splice-Switching ASO Targeting SMN2 Pre-mRNA: Nusinersen
4. Duchenne Muscular Dystrophy (DMD)
4.1. Clinical Manifestations and Genetic Background
4.2. Available Gene Therapies for DMD
4.2.1. GRT in DMD
GRTs Encoding Shortened DMD Genes: Microdystrophin GRTs
A GRT Encoding the GALGT2 Enzyme: rAAVrh74.MCK.GALGT2
4.2.2. ASOs in DMD
A Splice-Switching ASO Targeting Exon 51 of the DMD Pre-mRNA: Eteplirsen
A Splice-Switching ASO Targeting Exon 53 of the DMD Pre-mRNA: Golodirsen
A Splice-Switching ASO Targeting Exon 45 of the DMD Pre-mRNA: Casimersen
A Splice-Switching ASO Targeting Exon 53 of the DMD Pre-mRNA: Viltolarsen
A Peptide-Conjugated, Splice-Switching ASO Targeting Exon 51 of the DMD Pre-mRNA: SRP-5051
A Splice-Switching ASO Targeting Exon 45 of the DMD Pre-mRNA: Renadirsen
A Splice-Switching Stereopure ASO Targeting Exon 51 of the DMD Pre-mRNA: Suvodirsen
A Splice-Switching ASO Targeting Exon 53 of the DMD Pre-mRNA: WVE-N531
A Splice-Switching ASO Targeting Exon 44 of the DMD Pre-mRNA: NS-089/NCNP-02
An snRNP-Incorporated, Splice-Switching ASO Targeting Exon 2 of the DMD Pre-mRNA: scAAV9.U7.ACCA
5. Limb-Girdle Muscular Dystrophy (LGMD)
5.1. Clinical Manifestations and Genetic Background
5.2. Available Gene Therapies for LGMD
5.2.1. GRT in LGMD
A Dual-Vector GRT for LGMD Type 2B Dysferlinopathy: AAV.DYSF.DV
A Human γ-Sarcoglycan GRT for LGMD2C Sarcoglycanopathy
A Human hSGCA GRT for LGMD2D Sarcoglycanopathy: rAAV1.tMCK.hSGCA
A Human hSGCA GRT for LGMD2D Sarcoglycanopathy: AAVrh74.tMCK.hSGCA
A Human SGCB GRT for LGMD Type 2E Sarcoglycanopathy: SRP-9003
A Human FKRP GRT for LGMD Type 2I: LION-101
A Human FKRP GRT for LGMD Type 2I: GNT0006
6. Myotonic Dystrophy (DM)
6.1. Clinical Manifestations and Genetic Background
6.2. Available Gene Therapies for DM
6.2.1. ASOs in DM
An ASO RNase H1-Mediated Inhibitor of DMPK mRNA: IONIS-DMPKRx
6.2.2. RNAi in DM
A siRNA Inhibitor of DMPK mRNA: AOC-1001
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| AGO2 | argonaute 2 |
| AIDS | acquired immunodeficiency syndrome |
| ALS | amyotrophic lateral sclerosis |
| ASO | antisense oligonucleotide |
| ATXN2 | ataxin-2 |
| BBB | blood–brain barrier |
| Cas | CRISP-associated |
| CNS | central nervous system |
| CPP | cell-penetrating peptide |
| CSF | cerebrospinal fluid |
| C9orf72 | chromosome 9 open reading frame 72 |
| DM | myotonic dystrophy |
| DMD | Duchenne muscular dystrophy |
| DMPK | dystrophia myotonic protein kinase |
| DNA | deoxyribonucleic acid |
| dsRNA | double-stranded RNA |
| EAP | expanded access program |
| fALS | familial ALS |
| FDA | Food and Drug Administration |
| FKRP | fukutin-related protein |
| FLTD-TDP | frontotemporal lobar degeneration with TDP-43 inclusions |
| FTD | frontotemporal dementia |
| FUS | fused in sarcoma |
| GRT | gene-replacement therapy |
| HGF | hepatocyte growth factor |
| ILI | isolated limb infusion |
| IM | intramuscular |
| ISS-N1 | intronic splicing silencer-N1 |
| IT | intrathecal |
| IV | intravenous |
| LGMD | limb-girdle muscular dystrophy |
| mRNA | messenger RNA |
| miRNA | microRNA |
| OLE | open-label extension |
| PMO | phosphorodiamidate morpholino oligomer |
| polyQ | polyglutamine |
| PS | phosphorothioate |
| RISC | RNA-induced silencing complex |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| SMA | spinal muscular atrophy |
| SMN | survival of motor neuron |
| siRNA | small interfering RNA |
| SOD1 | superoxide dismutase 1 |
| TARDBP | TAR DNA-binding protein |
| TDP-43 | transactive response DNA-binding protein 43 |
| tMCK | muscle-specific creatine kinase (promoter) |
| UTR | untranslated region |
| vg | viral genome |
| 2′-O-Me | 2′-O-Methyl |
| 2′-O-MOE | 2′-O-methoxy ethyl/phosphorothioate |
References
- Verma, I.M.; Somia, N. Gene therapy-promises, problems and prospects. Nature 1997, 389, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hu, Y.; Ju, D. Gene therapy for neurodegenerative disorders: Advances, insights and prospects. Acta Pharm. Sin. B 2020, 10, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Aguti, S.; Malerba, A.; Zhou, H. The progress of AAV-mediated gene therapy in neuromuscular disorders. Expert Opin. Biol. Ther. 2018, 18, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Enoki, T.; Kawano, Y.; Veraz, M.; Nakai, H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat. Commun. 2014, 5, 3075. [Google Scholar] [CrossRef]
- Albright, B.H.; Storey, C.M.; Murlidharan, G.; Rivera, R.M.C.; Berry, G.E.; Madigan, V.J.; Asokan, A. Mapping the Structural Determinants Required for AAVrh.10 Transport across the Blood-Brain Barrier. Mol. Ther. 2018, 26, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Duque, S.; Joussemet, B.; Riviere, C.; Marais, T.; Dubreil, L.; Douar, A.-M.; Fyfe, J.; Moullier, P.; Colle, M.-A.; Barkats, M. Intravenous Administration of Self-complementary AAV9 Enables Transgene Delivery to Adult Motor Neurons. Mol. Ther. 2009, 17, 1187–1196. [Google Scholar] [CrossRef]
- Rafii, M.S.; Tuszynski, M.H.; Thomas, R.G.; Barba, D.; Brewer, J.B.; Rissman, R.A.; Siffert, J.; Aisen, P.S.; Mintzer, J.; Lerner, A.; et al. Adeno-Associated Viral Vector (Serotype 2)-Nerve Growth Factor for Patients With Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 834–841. [Google Scholar] [CrossRef]
- Au, H.K.E.; Isalan, M.; Mielcarek, M. Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 2022, 8, 809118. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Glazier, D.A.; Liao, J.; Roberts, B.L.; Li, X.; Yang, K.; Stevens, C.M.; Tang, W. Chemical Synthesis and Biological Application of Modified Oligonucleotides. Bioconjugate Chem. 2020, 31, 1213–1233. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Meena; Standley, S.M.; Lu, G.; Apponi, L.H.; et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef]
- Østergaard, M.E.; De Hoyos, C.L.; Wan, W.B.; Shen, W.; Low, A.; Berdeja, A.; Vasquez, G.; Murray, S.; Migawa, M.T.; Liang, X.-H.; et al. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020, 48, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Lundin, K.E.; Gissberg, O.; Smith, C.I.E.; Zain, R. Chemical Development of Therapeutic Oligonucleotides. Methods Mol. Biol. 2019, 2036, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The powerful world of anti-sense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Caplen, N.J.; Parrish, S.; Imani, F.; Fire, A.; Morgan, R.A. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl. Acad. Sci. USA 2001, 98, 9742–9747. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Rogers, S. Shope papilloma virus: A Passenger in man and its significance to the potential control of the host genome. Nature 1966, 212, 1220–1222. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangoul, H.; Bobruff, Y.; Cappellini, M.D.; Corbacioglu, S.; Fernandez, R.C.M.; De La Fuente, J.; Grupp, S.A.; Handgretinger, R.; Ho, T.W.; Imren, S.; et al. Safety and Efficacy of CTX001 in Patients with Transfusion-Dependent β-Thalassemia and Sickle Cell Disease: Early Results from the Climb THAL-111 and Climb SCD-121 Studies of Autologous CRISPR-CAS9-Modified CD34+ Hematopoietic Stem and Progenitor Cells. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Rosell, R.; Filipska, M.; Chaib, I.; Lligé, D.; Laguia, F. Commentary: Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Front. Oncol. 2020, 10, 1726. [Google Scholar] [CrossRef]
- Mullard, A. First in vivo CRISPR candidate enters the clinic. Nat. Rev. Drug Discov. 2019, 18, 656. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Nelson, C.E.; Robinson-Hamm, J.N.; Gersbach, C.A. Genome engineering: A new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol. 2017, 13, 647–661. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector–mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Yan, Z.; McCray, P.B., Jr.; Engelhardt, J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019, 28, R88–R94. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadou, E.; Seto, A.G.; Beatty, X.; Hermreck, M.; Gilles, M.-E.; Stroopinsky, D.; Pinter-Brown, L.C.; Pestano, L.; Marchese, C.; Avigan, D.; et al. Cobomarsen, an Oligonucleotide Inhibitor of miR-155, Slows DLBCL Tumor Cell Growth In Vitro and In Vivo. Clin. Cancer Res. 2021, 27, 1139–1149. [Google Scholar] [CrossRef]
- Gilboa, E.; Smith, C. Gene therapy for infectious diseases: The AIDS model. Trends Genet. 1994, 10, 139–144. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Writing Group on behalf of the Edaravone (MCI-186) ALS 17 Study Group. Exploratory double-blind, par-allel-group, placebo-controlled extension study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 2006, 7, 247–251. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V.; The ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic Lateral sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Bellingham, M.C. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: What have we learned in the last decade? CNS Neurosci. Ther. 2011, 17, 4–31. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.; Hwu, W.-L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Nigel Leigh, P. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Phukan, J.; Pender, N.; Hardiman, O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007, 6, 994–1003. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef]
- Pyun, W.-B.; Hahn, W.; Kim, D.-S.; Yoo, W.-S.; Lee, S.-D.; Won, J.-H.; Rho, B.-S.; Park, Z.-Y.; Kim, J.-M.; Kim, S. Naked DNA expressing two isoforms of hepatocyte growth factor induces collateral artery augmentation in a rabbit model of limb ischemia. Gene Ther. 2010, 17, 1442–1452. [Google Scholar] [CrossRef]
- Taniyama, Y.; Morishita, R.; Hiraoka, K.; Aoki, M.; Nakagami, H.; Yamasaki, K.; Matsumoto, K.; Nakamura, T.; Kaneda, Y.; Ogihara, T. Therapeutic angiogenesis induced by human hepatocyte growth factor gene in rat diabetic hind limb Ischemia model. Circulation 2001, 104, 2344–2350. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Guo, G.F.; Martinez, J.A.; Singh, V.; Zochodne, D.W. Dynamic plasticity of axons within a cutaneous milieu. J. Neurosci. 2010, 30, 14735–14744. [Google Scholar] [CrossRef] [PubMed]
- Ebens, A.; Brose, K.; Leonardo, E.D.; Hanson, M.G., Jr.; Bladt, F.; Birchmeier, C.; Barres, B.A.; Tessier-Lavigne, M. Hepatocyte Growth Factor/Scatter Factor Is an Axonal Chemoattractant and a Neurotrophic Factor for Spinal Motor Neurons. Neuron 1996, 17, 1157–1172. [Google Scholar] [CrossRef] [Green Version]
- Funakoshi, H.; Nakamura, T. Identification of HGF-like protein as a novel neurotrophic factor for avian dorsal root ganglion sensory neurons. Biochem. Biophys. Res. Commun. 2001, 283, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Gaillard, S.; Malapert, P.; Liu, Y.; Rodat-Despoix, L.; Samokhvalov, I.M.; Delmas, P.; Helmbacher, F.; Maina, F.; Moqrich, A. Hepatocyte growth factor-met signaling is required for Runx1 extinction and peptidergic differentiation in Primary nociceptive neurons. J. Neurosci. 2010, 30, 12414–12423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, N.; Yamanaka, H.; Fukuoka, T.; Dai, Y.; Obata, K.; Mashimo, T.; Noguchi, K. Expression of HGF and cMet in the peripheral nervous system of adult rats following sciatic nerve injury. NeuroReport 2001, 12, 1403–1407. [Google Scholar] [CrossRef]
- Kato, N.; Nemoto, K.; Nakanishi, K.; Morishita, R.; Kaneda, Y.; Uenoyama, M.; Ikeda, T.; Fujikawa, K. Nonviral HVJ (hemagglutinating virus of Japan) liposome-mediated retrograde gene transfer of human hepatocyte growth factor into rat nervous system promotes functional and histological recovery of the crushed nerve. Neurosci. Res. 2005, 52, 299–310. [Google Scholar] [CrossRef]
- Ko, K.R.; Lee, J.; Lee, D.; Nho, B.; Kim, S. Hepatocyte Growth Factor (HGF) Promotes Peripheral Nerve Regeneration by Activating Repair Schwann Cells. Sci. Rep. 2018, 8, 8316. [Google Scholar] [CrossRef] [Green Version]
- Maina, F.; Hilton, M.C.; Ponzetto, C.; Davies, A.M.; Klein, R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997, 11, 3341–3350. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.; Glass, D.J.; Arriaga, R.; Yancopoulos, G.D.; Lindsay, R.M.; Conn, G. Hepatocyte growth factor promotes motor neuron survival and synergizes with ciliary neurotrophic factor. J. Biol. Chem. 1997, 272, 5187–5191. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-M.; Toma, J.G.; Bamji, S.X.; Belliveau, D.J.; Kohn, J.; Park, M.; Miller, F.D. Autocrine hepatocyte growth factor provides a local mechanism for promoting axonal growth. J. Neurosci. 1998, 18, 8369–8381. [Google Scholar] [CrossRef] [Green Version]
- Kessler, J.A.; Shaibani, A.; Sang, C.N.; Christiansen, M.; Kudrow, D.; Vinik, A.; Shin, N. Gene therapy for diabetic peripheral neuropathy: A randomized, placebo-controlled phase III study of VM202, a plasmid DNA encoding human hepatocyte growth factor. Clin. Transl. Sci. 2021, 14, 1176–1184. [Google Scholar] [CrossRef]
- Kibbe, M.R.; Hirsch, A.T.; Mendelsohn, F.O.; Davies, M.G.; Pham, H.; Saucedo, J.; Marston, W.; Pyun, W.-B.; Min, S.-K.; Peterson, B.G.; et al. Safety and efficacy of plasmid DNA expressing two isoforms of hepatocyte growth factor in patients with critical limb ischemia. Gene Ther. 2015, 23, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Helixmith. Available online: https://www.helixmith.com/bbs/board.php?bo_table=s5_1_eng&wr_id=36 (accessed on 27 March 2022).
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.; Pestronk, P.A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. A Phase I, Randomised, First-in-Human Study of an Antisense Oligonucleotide Directed Against SOD1 Delivered Intrathecally in SOD1-Familial ALS Patients. Lancet Neurol. 2013, 12, 435. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Biogen. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its (accessed on 27 March 2022).
- Fernandez, M.; McClain, M.E.; Martinez, R.A.; Snow, K.; Lipe, H.; Ravits, J.; Bird, T.D.; La Spada, A.R. Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology 2000, 55, 569–572. [Google Scholar] [CrossRef]
- Wang, M.-D.; Gomes, J.; Cashman, N.R.; Little, J.; Krewski, D. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS−A systematic review and meta-analysis of observational studies. PLoS ONE 2014, 9, e105534. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Correction: Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and Pathological brain tissue. PLoS ONE 2010, 5, e13205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Daigle, J.G.; Cunningham, K.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Saánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dodart, J.-C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef]
- Liu, Y.; Andreucci, A.; Iwamoto, N.; Yin, Y.; Yang, H.; Liu, F.; Patil, S.; Mohapatra, S.; Purcell-Estabrook, E.; Taborn, K.; et al. WVE-004, an investigational stereopure antisense oligonucleotide for the treatment of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (2302). Neurology 2021, 96 (Suppl. 15), 2302. [Google Scholar]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of Poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C. Tailored treatment for ALS poised to move ahead. Nat. Med. 2019; ahead of print. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Cabrera, G.T.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1G93A Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H.; Mueller, C. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med. 2018, 10, eaau6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H., Jr.; Sena-Esteves, M. Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Castro, D.; Iannaccone, S.T. Spinal Muscular Atrophy: Therapeutic Strategies. Curr. Treat. Options Neurol. 2014, 16, 316. [Google Scholar] [CrossRef]
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.; Turner, B.; Gillingwater, T.; Swoboda, K.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2017, 81, 355–368. [Google Scholar] [CrossRef]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Day, J.W.; Darras, B.T.; Kuntz, N.L.; Connolly, A.M.; Crawford, T.; Mendell, J.R. One-Time Intrathecal (IT) Administration of AVXS-101 IT Gene-Replacement Therapy for Spinal Muscular Atrophy: Phase 1 Study (STRONG) (2493). Neurology 2020, 94 (Suppl. 15), 2493. [Google Scholar]
- Strauss, K.; Farrar, M.; Muntoni, F.; Saito, K.; Mendell, J.; Servais, L.; McMillan, H.; Finkel, R.; Swoboda, K.; Kwon, J.; et al. OPR-201 Onasemnogene abeparvovec for presymptomatic infants with spinal muscular atrophy and 2 copies of SMN2: A phase III study. Eur. J. Neurol. 2021, 28 (Suppl. S1), S950–S951. [Google Scholar]
- Strauss, K.A.; Muntoni, F.; Farrar, M.A.; Saito, K.; Mendell, J.; Servais, L.; Macek, T. Onasemnogene abeparvovec gene therapy in presymptomatic spinal muscular atrophy (SMA): SPR1NT study update in children with 3 copies of SMN2 (4163). Neurology 2021, 96 (Suppl. 15), 4163. [Google Scholar]
- Dangouloff, T.; Vrščaj, E.; Servais, L.; Osredkar, D.; Adoukonou, T.; Aryani, O.; Barisic, N.; Bashiri, F.; Bastaki, L.; Benitto, A.; et al. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul. Disord. 2021, 31, 574–582. [Google Scholar] [CrossRef]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Haché, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy (accessed on 27 March 2022).
- Darras, B.T.; Farrar, M.A.; Mercuri, E.; Finkel, R.S.; Foster, R.; Hughes, S.G.; Bhan, I.; Farwell, W.; Gheuens, S. An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs 2019, 33, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Cecio, M.R.; Torre, V.; De Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizers-new-phase-1b-results-gene-therapy-ambulatory-boys (accessed on 27 March 2022).
- Crudele, J.M.; Chamberlain, J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019, 28, R102–R107. [Google Scholar] [CrossRef] [Green Version]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; Dealwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Kesselheim, A.; Avorn, J. Approving a Problematic Muscular Dystrophy Drug: Implications for FDA Policy. JAMA 2016, 316, 2357–2358. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study Group and Telethon Foundation DMD Italian Network. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [Green Version]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef]
- Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Egawa, Y.; Takeda, S. DMD CLINICAL THERAPIES II: P. 129A Japanese phase I/II study of NS-065/NCNP-01, exon 53 skipping drug, in patients with Duchenne muscular dystrophy—A dose-finding study. Neuromuscul. Disord. 2018, 28, S68. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation (accessed on 27 March 2022).
- NS Pharma. Available online: https://www.nspharma.com/pdfs/NSPharma_Long-term_Data_PPMD_New.pdf (accessed on 27 March 2022).
- Amantana, A.; Iversen, P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005, 5, 550–555. [Google Scholar] [CrossRef]
- Gebski, B.L.; Mann, C.J.; Fletcher, S.; Wilton, S.D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 2003, 12, 1801–1811. [Google Scholar] [CrossRef]
- Richard, J.-P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sarepta Therapeutics. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-positive-clinical-results (accessed on 27 March 2022).
- Ito, K.; Takakusa, H.; Kakuta, M.; Kanda, A.; Takagi, N.; Nagase, H.; Watanabe, N.; Asano, D.; Goda, R.; Masuda, T.; et al. Renadirsen, a Novel 2′OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo. Curr. Issues Mol. Biol. 2021, 43, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Daiichi Sankyo. Available online: https://www.daiichisankyo.com/media/press_release/detail/index_4112.html (accessed on 27 March 2022).
- Wagner, K.; Phan, H.; Servais, L.; Gidaro, T.; Cripe, L.; Eagle, M.; Muntoni, F.; Straub, V.; Lonkar, P.; Schmitz, S.; et al. O.28Safety and tolerability of suvodirsen (WVE-210201) in patients with Duchenne muscular dystrophy: Results from a phase 1 clinical trial. Neuromuscul. Disord. 2019, 29, S124. [Google Scholar] [CrossRef]
- Wave Life Sciences. Available online: https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-discontinuation-suvodirsen (accessed on 27 March 2022).
- Wave Life Sciences. Available online: https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-initiation-dosing-phase-1b2a (accessed on 27 March 2022).
- Gadgil, A.; Raczyńska, K.D. U7 snRNA: A tool for gene therapy. J. Gene Med. 2021, 23, e3321. [Google Scholar] [CrossRef]
- Walton, J.N.; Nattrass, F.J. On the classification, natural history and treatment of the myopathies. Brain 1954, 77, 169–231. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.M.D. Limb-girdle muscular dystrophy. In Diagnostic Criteria for Neuromuscular Disorders; Emery, A.E.H., Ed.; ENMC: Baarn, The Netherlands, 1994; pp. 25–31. [Google Scholar]
- Bushby, K.; Muntoni, F.; Bourke, J.P. 107th ENMC International Workshop: The management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th–9th June 2002, Naarden, the Netherlands. Neuromuscul. Disord. 2003, 13, 166–172. [Google Scholar] [CrossRef]
- Kirschner, J.; Bönnemann, C.G. The congenital and limb-girdle muscular dystrophies: Sharpening the focus, blurring the boundaries. Arch. Neurol. 2004, 61, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Minetti, C.; Sotgia, F.; Bruno, C.; Scartezzini, P.; Broda, P.; Bado, M.; Masetti, E.; Mazzocco, M.; Egeo, A.; Donati, M.A.; et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998, 18, 365–368. [Google Scholar] [CrossRef]
- Narayanaswami, P.; Weiss, M.; Selcen, D.; David, W.; Raynor, E.; Carter, G.; Wicklund, M.; Barohn, R.J.; Ensrud, E.; Griggs, R.; et al. Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014, 83, 1453–1463. [Google Scholar] [CrossRef]
- Wicklund, M.P.; Kissel, J.T. The Limb-Girdle Muscular Dystrophies. Neurol. Clin. 2014, 32, 729–749. [Google Scholar] [CrossRef]
- Iyadurai, S.J.P.; Kissel, J.T. The Limb-Girdle Muscular Dystrophies and the Dystrophinopathies. Contin. Lifelong Learn. Neurol. 2016, 22, 1954–1977. [Google Scholar] [CrossRef]
- Liewluck, T.; Milone, M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve 2018, 58, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Pajusalu, S.; Lake, N.J.; Zhou, G.; Ioannidis, N.; Mittal, P.; Johnson, N.E.; Weihl, C.C.; Williams, B.A.; Albrecht, D.E.; et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet. Med. 2019, 21, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998, 20, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Ben Hamida, M.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Illa, I.; Serrano-Munuera, C.; Gallardo, E.; Lasa, A.; Rojas-García, R.; Palmer, J.; Gallano, P.; Baiget, M.; Matsuda, C.; Brown, R.H. Distal anterior compartment myopathy: A dysferlin mutation causing a new muscular dystrophy phenotype. Ann. Neurol. 2001, 49, 130–134. [Google Scholar] [CrossRef]
- Paradas, C.; Llauger, J.; Diaz-Manera, J.; Rojas-Garcia, R.; De Luna, N.; Iturriaga, C.; Marquez, C.; Uson, M.; Hankiewicz, K.; Gallardo, E.; et al. Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology 2010, 75, 316–323. [Google Scholar] [CrossRef]
- Nguyen, K.; Bassez, G.; Krahn, M.; Bernard, R.; Laforêt, P.; Labelle, V.; Urtizberea, J.A.; Figarella-Branger, D.; Romero, N.; Attarian, S.; et al. Phenotypic study in 40 patients with dysferlin gene mutations: High frequency of atypical phenotypes. Arch. Neurol. 2007, 64, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Ueyama, H.; Kumamoto, T.; Horinouchi, H.; Fujimoto, S.; Aono, H.; Tsuda, T. Clinical Heterogeneity in Dysferlinopathy. Intern. Med. 2002, 41, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Pozsgai, E.; Griffin, D.; Potter, R.; Sahenk, Z.; Lehman, K.; Rodino-Klapac, L.R.; Mendell, J.R. Unmet needs and evolving treatment for limb girdle muscular dystrophies. Neurodegener. Dis. Manag. 2021, 11, 411–429. [Google Scholar] [CrossRef]
- Weiler, T.; Bashir, R.; Anderson, L.V.B.; Davison, K.; Moss, J.A.; Britton, S.; Nylen, E.; Keers, S.; Vafiadaki, E.; Greenberg, C.R.; et al. Identical Mutation in Patients with Limb Girdle Muscular Dystrophy Type 2B Or Miyoshi Myopathy Suggests a Role for Modifier Gene(s). Hum. Mol. Genet. 1999, 8, 871–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueyama, H.; Kumamoto, T.; Nagao, S.-I.; Masuda, T.; Horinouchi, H.; Fujimoto, S.; Tsuda, T. A new dysferlin gene mutation in two Japanese families with limb-girdle muscular dystrophy 2B and Miyoshi myopathy. Neuromuscul. Disord. 2001, 11, 139–145. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Numitone, G.; Aurino, S.; Caserta, I.R.; Fanin, M.; Politano, L.; Minetti, C.; Ricci, E.; Piluso, G.; Angelini, C.; et al. Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur. J. Hum. Genet. 2011, 19, 974–980. [Google Scholar] [CrossRef]
- Sondergaard, P.C.; Griffin, D.A.; Pozsgai, E.R.; Johnson, R.W.; Grose, W.E.; Heller, K.N.; Shontz, K.M.; Montgomery, C.L.; Liu, J.; Clark, K.R.; et al. AAV. Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015, 2, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.A.; Griffin, D.A.; Sondergaard, P.C.; Johnson, R.W.; Pozsgai, E.R.; Heller, K.N.; Peterson, E.L.; Lehtimäki, K.K.; Windish, H.P.; Mittal, P.J.; et al. Systemic Delivery of Dysferlin Overlap Vectors Provides Long-Term Gene Expression and Functional Improvement for Dysferlinopathy. Hum. Gene Ther. 2018, 29, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, L.E.; Duclos, F.; Broux, O.; Bourg, N.; Sunada, Y.; Allamand, V.; Meyer, J.; Richard, I.; Moomaw, C.; Slaughter, C.; et al. β–sarcoglycan: Characterization and role in limb–girdle muscular dystrophy linked to 4q12. Nat. Genet. 1995, 11, 257–265. [Google Scholar] [CrossRef]
- Noguchi, S.; McNally, E.M.; Ben Othmane, K.; Hagiwara, Y.; Mizuno, Y.; Yoshida, M.; Yamamoto, H.; Bönnemann, C.G.; Gussoni, E.; Denton, P.H.; et al. Mutations in the Dystrophin-Associated Protein γ-Sarcoglycan in Chromosome 13 Muscular Dystrophy. Science 1995, 270, 819–822. [Google Scholar] [CrossRef]
- Duggan, D.J.; Gorospe, J.R.; Fanin, M.; Hoffman, E.P.; Angelini, C.; Pegoraro, E.; Noguchi, S.; Ozawa, E.; Pendlebury, W.; Waclawik, A.; et al. Mutations in the Sarcoglycan Genes in Patients with Myopathy. N. Engl. J. Med. 1997, 336, 618–625. [Google Scholar] [CrossRef]
- Vainzof, M.; Souza, L.S.; Gurgel-Giannetti, J.; Zatz, M. Sarcoglycanopathies: An update. Neuromuscul. Disord. 2021, 31, 1021–1027. [Google Scholar] [CrossRef]
- Herson, S.; Hentati, F.; Rigolet, A.; Behin, A.; Romero, N.B.; Leturcq, F.; Laforêt, P.; Maisonobe, T.; Amouri, R.; Haddad, H.; et al. A phase I trial of adeno-associated virus serotype 1-γ-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012, 135, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1–12. [Google Scholar] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Rosales-Quintero, X.; Kota, J.; Coley, B.; Galloway, G.; Craenen, J.M.; Lewis, S.; Malik, V.; Ms, C.S.; et al. Limb-girdle muscular dystrophy type 2D gene therapy restores α-sarcoglycan and associated proteins. Ann. Neurol. 2009, 66, 290–297. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Rosales, X.Q.; Coley, B.; Galloway, G.; Lewis, S.L.; Malik, V.; Shilling, C.; Byrne, B.J.; Conlon, T.J.; et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 2010, 68, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Chicoine, L.G.; Al-Zaidy, S.A.; Sahenk, Z.; Lehman, K.; Lowes, L.; Miller, N.; Alfano, L.; Galliers, B.; Lewis, S.; et al. Gene Delivery for Limb-Girdle Muscular Dystrophy Type 2D by Isolated Limb Infusion. Hum. Gene Ther. 2019, 30, 794–801. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Passos-Bueno, M.R.; McNally, E.M.; Vainzof, M.; Moreira, E.D.S.; Marie, S.; Pavanello, R.C.M.; Noguchi, S.; Ozawa, E.; Zatz, M.; et al. Genomic screening for beta-sarcoglycan gene mutations: Missense mutations may cause severe limb-girdle muscular dystrophy type 2E (LGMD 2E). Hum. Mol. Genet. 1996, 5, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semplicini, C.; Vissing, J.; Dahlqvist, J.R.; Stojkovic, T.; Bello, L.; Witting, N.; Duno, M.; Leturcq, F.; Bertolin, C.; D’Ambrosio, P.; et al. Clinical and genetic spectrum in limb-girdle muscular dystrophy type 2E. Neurology 2015, 84, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Pozsgai, E.R.; Griffin, D.A.; Heller, K.N.; Mendell, J.R.; Rodino-Klapac, L.R. Systemic AAV-Mediated β-Sarcoglycan Delivery Targeting Cardiac and Skeletal Muscle Ameliorates Histological and Functional Deficits in LGMD2E Mice. Mol. Ther. 2017, 25, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Sarepta Therapeutics. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-investigational-gene-therapy-srp-9003-0 (accessed on 27 March 2022).
- Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Manya, H.; Kuga, A.; Yamaguchi, Y.; Akasaka-Manya, K.; Furukawa, J.-I.; Mizuno, M.; Kawakami, H.; et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. 2016, 14, 2209–2223. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Brockington, M.; Straub, V.; Quijano-Roy, S.; Yuva, Y.; Herrmann, R.; Brown, S.; Torelli, S.; Dubowitz, V.; Blake, D.J.; et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann. Neurol. 2003, 53, 537–542. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Harley, H.G.; Brook, J.D.; Rundle, S.A.; Crow, S.; Reardon, W.; Buckler, A.J.; Harper, P.S.; Housman, D.E.; Shaw, D.J. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 1992, 355, 545–546. [Google Scholar] [CrossRef] [PubMed]
- Aslanidis, C.; Jansen, G.; Amemiya, C.; Shutler, G.; Mahadevan, M.; Tsilfidis, C.; Chen, C.; Alleman, J.; Wormskamp, N.G.M.; Vooijs, M.; et al. Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature 1992, 355, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Kaliman, P.; Llagostera, E. Myotonic dystrophy protein kinase (DMPK) and its role in the pathogenesis of myotonic dystrophy 1. Cell. Signal. 2008, 20, 1935–1941. [Google Scholar] [CrossRef]
- Ho, T.H.; Charlet-B, N.; Poulos, M.G.; Singh, G.; Swanson, M.S.; Cooper, T.A. Muscleblind proteins regulate al-ternative splicing. EMBO J. 2004, 23, 3103–3112. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef] [Green Version]
- Mulders, S.A.M.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.E.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.; Yokota, T. Degradation of Toxic RNA in Myotonic Dystrophy Using Gapmer Antisense Oligonucleotides. Methods Mol. Biol. 2020, 2176, 99–109. [Google Scholar] [CrossRef]
- Wang, E.T.; Cody, N.A.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [Green Version]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.; Thornton, C.A. Expanded CUG Repeats Trigger Aberrant Splicing of ClC-1 Chloride Channel Pre-mRNA and Hyperexcitability of Skeletal Muscle in Myotonic Dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Lueck, J.D.; Mankodi, A.; Swanson, M.S.; Thornton, C.A.; Dirksen, R.T. Muscle Chloride Channel Dysfunction in Two Mouse Models of Myotonic Dystrophy. J. Gen. Physiol. 2007, 129, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.A.; Moxley, R.T.; Day, J.W.; Ashizawa, T.; Barohn, R.; Leung, D.; Kissel, J.; Johnson, N.; Eichinger, K.; Hardage, J.; et al. Study Design of a Phase 1/2a Trial with ISIS-DMPKRx for the Treatment of Myotonic Dystrophy Type 1 (P3.167). Neurology 2016, 86 (Suppl. 16), P3.167. [Google Scholar]
- Mignon, L.; Norris, D.; Bishop, K.; Derosier, F.; LANE, R.; Bennett, F. ISIS-DMPKRx in Healthy Volunteers: A Placebo-controlled, Randomized, Single Ascending-Dose Phase 1 Study (P3.166). Neurology 2016, 86 (Suppl. 16), P3.166. [Google Scholar]
- Hu, N.; Antoury, L.; Baran, T.M.; Mitra, S.; Bennett, C.F.; Rigo, F.; Foster, T.; Wheeler, T.M. Non-invasive monitoring of alternative splicing outcomes to identify candidate therapies for myotonic dystrophy type 1. Nat. Commun. 2018, 9, 5227. [Google Scholar] [CrossRef]
- Avidity Biosciences. Available online: https://aviditybiosciences.investorroom.com/2021-08-02-Avidity-Biosciences-Receives-IND-Clearance-from-FDA-to-Proceed-with-the-Phase-1-2-MARINA-TM-Trial-of-AOC-1001-in-Adults-with-Myotonic-Dystrophy-DM1] (accessed on 27 March 2022).
- Avidity Biosciences. Available online: https://aviditybiosciences.investorroom.com/2021-11-04-Avidity-Announces-First-Person-Dosed-with-an-Antibody-Oligonucleotide-Conjugate-AOC-TM (accessed on 27 March 2022).
- Brenner, D.; Ludolph, A.C.; Weishaupt, J.H. Gene specific therapies—The next therapeutic milestone in neurology. Neurol. Res. Pract. 2020, 2, 25. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Hordeaux, J.; Buza, E.L.; Dyer, C.; Goode, T.; Mitchell, T.W.; Richman, L.; Denton, N.; Hinderer, C.; Katz, N.; Schmid, R.; et al. Adeno-Associated Virus-Induced Dorsal Root Ganglion Pathology. Hum. Gene Ther. 2020, 31, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, Y. Innate Immune Recognition of Viruses and Viral Vectors. Hum. Gene Ther. 2009, 20, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Sands, M.S.; Venditti, C.P. Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum. Gene Ther. 2017, 28, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Finkel, R.; Mercuri, E.; Muntoni, F. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc. Health 2018, 2, 600–609. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347, Erratum in Nat. Med. 2006, 12, 592. [Google Scholar] [CrossRef]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing Anti–Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Samaranch, L.; Salegio, E.A.; San Sebastian, W.; Kells, A.P.; Foust, K.D.; Bringas, J.R.; Lamarre, C.; Forsayeth, J.; Kaspar, B.K.; Bankiewicz, K.S. Adeno-Associated Virus Serotype 9 Transduction in the Central Nervous System of Nonhuman Primates. Hum. Gene Ther. 2012, 23, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.J.; Nagabhushan Kalburgi, S.; McCown, T.J.; Jude Samulski, R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013, 20, 450–459. [Google Scholar] [CrossRef] [Green Version]
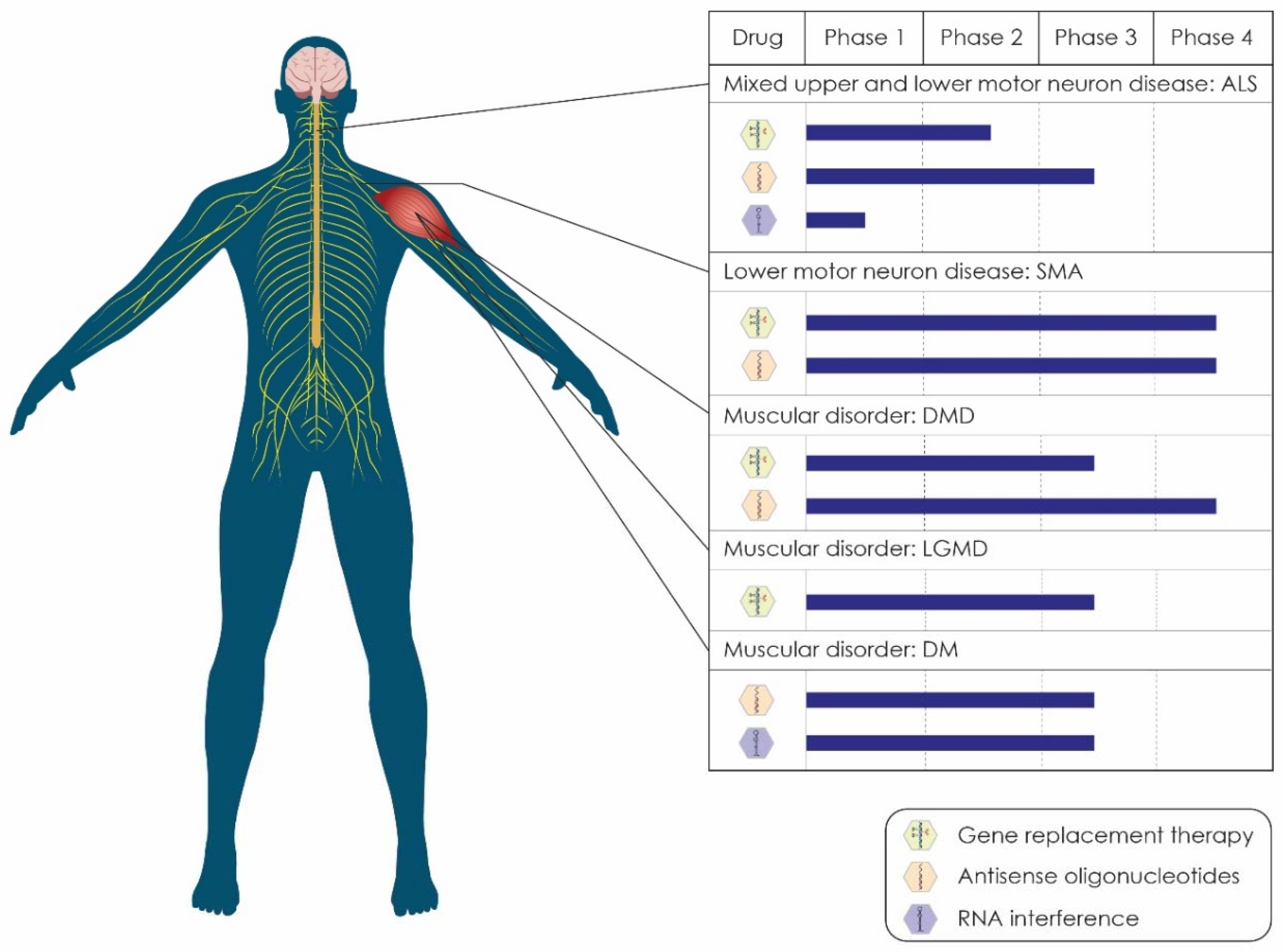

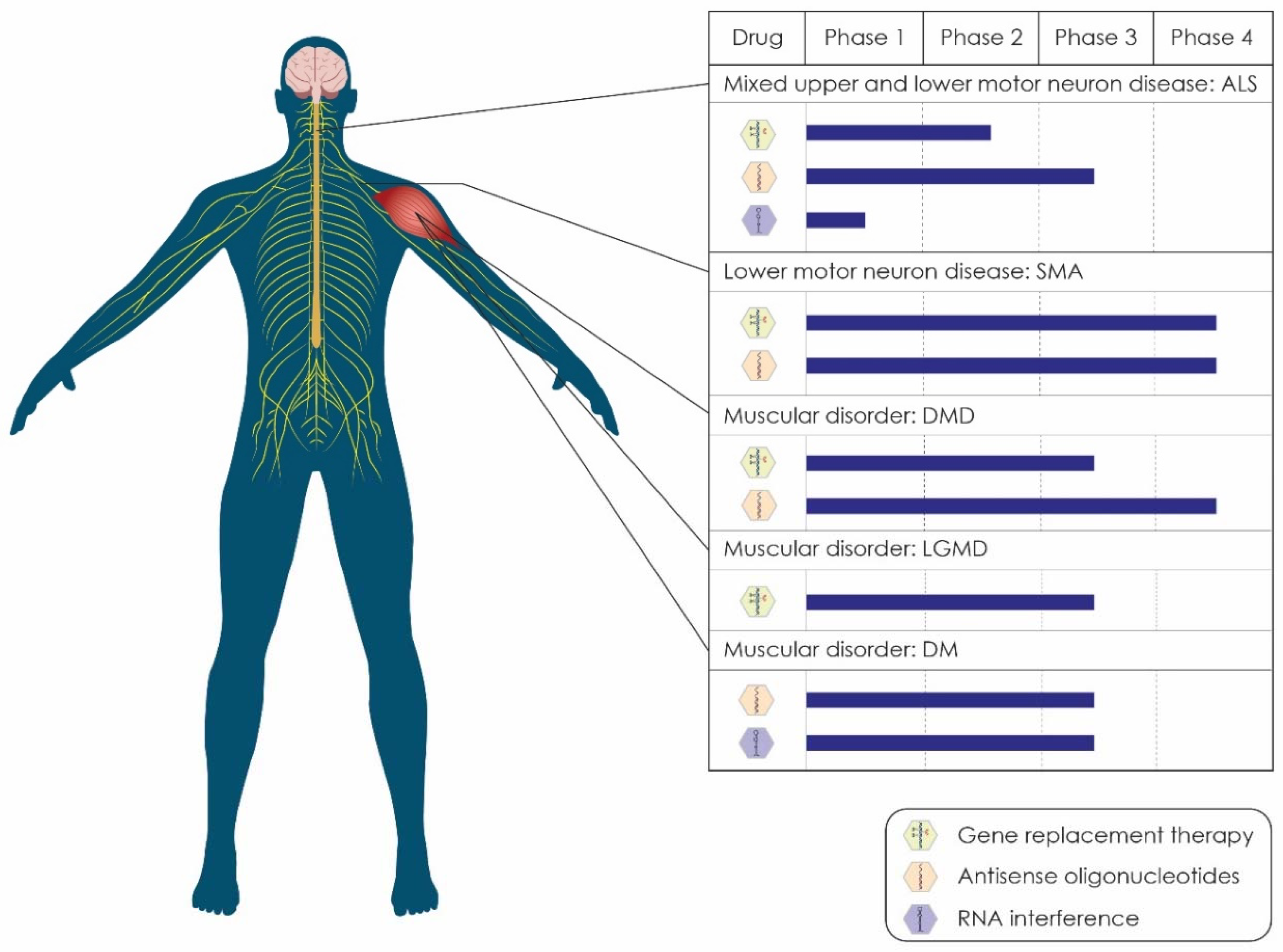{kind=link}
| Drug | Sponsor/Collaborators | Target of Drug | Status | NCT Number | Study Completion | |
|---|---|---|---|---|---|---|
| Gene replacement therapies (GRTs) | ||||||
| 1 | Engensis | Helixmith | GRT of HGF gene | Phase 2 | NCT05176093 | November 2022 |
| 2 | Engensis | Helixmith | GRT of HGF gene | Phase 2 | NCT04632225 | July 2022 |
| Antisense oligonucleotides (ASOs) | ||||||
| 1 | BIIB078 | Biogen | ASO that targets C9ORF72 mRNA | Phase 1 | NCT04288856 | July 2023 |
| 2 | BIIB105 | Ionis/Biogen | ASO that targets ATXN2 mRNA | Phase 1 | NCT04494256 | December 2024 |
| 3 | WVE-004 | Wave Life Sciences | ASO that targets C9orf72 mRNA | Phase 1/2 | NCT04931862 | February 2023 |
| 4 | Jacifusen | Ionis | ASO that targets FUS mRNA | Phase 3 | NCT04768972 | March 2024 |
| 5 | Tofersen | Ionis/Biogen | ASO that targets SOD1 mRNA | Phase 3 | NCT03070119 | June 2024 |
| 6 | Tofersen | Biogen | ASO that targets SOD1 mRNA | Phase 3 | NCT04856982 | August 2027 |
| Drug | Sponsor/Collaborators | Target of Drug | Status | NCT Number | Study Completion | |
|---|---|---|---|---|---|---|
| Gene replacement therapies (GRTs) | ||||||
| 1 | Zolgensma | Novartis | AAV-based GRT of SMN gene | Phase 3 | NCT04851873 | August 2023 |
| 2 | Zolgensma | Novartis | AAV-based GRT of SMN gene | Phase 3 | NCT05089656 | October 2024 |
| 3 | Zolgensma | Novartis | AAV-based GRT of SMN gene | Phase 4 | NCT05073133 | May 2023 |
| 4 | Zolgensma | Novartis | AAV-based GRT of SMN gene | Phase 4 | NCT04042025 | December 2035 |
| Antisense oligonucleotides (ASOs) | ||||||
| 1 | Nusinersen | Biogen | ASO that targets SMN2 mRNA | Phase 2 | NCT02386553 | January 2025 |
| 2 | Nusinersen | Biogen | ASO that targets SMN2 mRNA | Phase 2/3 | NCT04089566 | July 2023 |
| 3 | Nusinersen | Biogen | ASO that targets SMN2 mRNA | Phase 3 | NCT02594124 | August 2023 |
| 4 | Nusinersen | Biogen | ASO that targets SMN2 mRNA | Phase 3 | NCT04729907 | May 2026 |
| 5 | Nusinersen | Biogen | ASO that targets SMN2 mRNA | Phase 3 | NCT05067790 | June 2027 |
| 6 | Nusinersen | Biogen | ASO that targets SMN2 mRNA | Phase 4 | NCT04488133 | September 2024 |
| 7 | Nusinersen | CHU de Reims | ASO that targets SMN2 mRNA | NA | NCT04576494 | May 2024 |
| 8 | Nusinersen | CHU de Nice | ASO that targets SMN2 mRNA | NA | NCT04159987 | November 2022 |
| 9 | Nusinersen | Policlinico Gemelli | ASO that targets SMN2 mRNA | NA | NCT04674618 | December 2022 |
| Drug | Sponsor/Collaborators | Target of Gene Therapy | Status | NCT Number | Study Completion | |
|---|---|---|---|---|---|---|
| Gene replacement therapies (GRTs) | ||||||
| 1 | SRP-9001 | Sarepta Therapeutics | GRT of micro-dystrophin gene | Phase 1 | NCT04626674 | July 2026 |
| 2 | PF-06939926 | Pfizer | GRT of micro-dystrophin gene | Phase 1 | NCT03362502 | May 2028 |
| 3 | rAAVrh74.MCK.GALGT2 | Nationwide Children’s Hospital | GRT of GALGT2 gene | Phase 1/2 | NCT03333590 | November 2021 |
| 4 | SRP-9001 | Sarepta Therapeutics | GRT of micro-dystrophin gene | Phase 1/2 | NCT03375164 | April 2023 |
| 5 | SGT-001 | Solid Biosciences | GRT of micro-dystrophin gene | Phase 1/2 | NCT03368742 | December 2028 |
| 6 | SRP-9001 | Sarepta Therapeutics | GRT of micro-dystrophin gene | Phase 3 | NCT05096221 | November 2024 |
| 7 | PF-06939926 | Pfizer | GRT of micro-dystrophin gene | Phase 3 | NCT04281485 | September 2028 |
| Antisense oligonucleotides (ASOs) | ||||||
| 1 | NS-089/NCNP-02 | Nippon Shinyaku | ASO that targets exon 44 of DMD mRNA | Phase 1/2 | NCT04129294 | December 2021 |
| 2 | WVE-N531 | Wave Life Sciences | ASO that targets exon 53 of DMD mRNA | Phase 1/2 | NCT04906460 | September 2022 |
| 3 | scAAV9.U7.ACCA | Megan Waldrop/Audentes Therapeutics | snRNA that targets exon 2 of DMD mRNA | Phase 1/2 | NCT04240314 | November 2025 |
| 4 | Eteplirsen | Sarepta Therapeutics | ASO that targets exon 51 of DMD mRNA | Phase 2 | NCT04179409 | September 2022 |
| 5 | DS-5141b | Daiichi Sankyo (Japan) | ASO that targets exon 45 of DMD mRNA | Phase 2 | NCT04433234 | March 2023 |
| 6 | Viltolarsen | NS Pharma | ASO that targets exon 53 of DMD mRNA | Phase 2 | NCT05135663 | July 2023 |
| 7 | Vitolarsen | NS Pharma | ASO that targets exon 53 of DMD mRNA | Phase 2 | NCT04956289 | May 2024 |
| 8 | SRP-5051 | Sarepta Therapeutics | ASO that targets exon 51 of DMD mRNA | Phase 2 | NCT04004065 | August 2024 |
| 9 | Eteplirsen | Sarepta Therapeutics | ASO that targets exon 51 of DMD mRNA | Phase 2 | NCT03985878 | February 2027 |
| 10 | Casimersen & Golodirsen | Sarepta Therapeutics | ASOs that target exon 45 & exon 53 of DMD mRNA | Phase 3 | NCT02500381 | April 2024 |
| 11 | Vitolarsen | NS Pharma | ASO that targets exon 53 of DMD mRNA | Phase 3 | NCT04060199 | December 2024 |
| 12 | Eteplirsen | Sarepta Therapeutics | ASO that targets exon 51 of DMD mRNA | Phase 3 | NCT03992430 | February 2026 |
| 13 | Vitolarsen | NS Pharma | ASO that targets exon 53 of DMD mRNA | Phase 3 | NCT04768062 | June 2026 |
| 14 | Casimersen & Golodirsen | Sarepta Therapeutics | ASOs that target exon 45 & exon 53 of DMD mRNA | Phase 3 | NCT03532542 | August 2026 |
| 15 | Vitolarsen | NS Pharma | ASO that targets exon 53 of DMD mRNA | Phase 4 | NCT04687020 | November 2031 |
| Drug | Sponsor/Collaborators | Target of Gene Therapy | Status | NCT Number | Study Completion | |
|---|---|---|---|---|---|---|
| 1 | SRP-9003 | Sarepta Therapeutics | GRT of SGCB gene | Phase 1/2 | NCT03652259 | February 2025 |
| 3 | LION-101 | Asklepios Biopharmaceutical | GRT of FKRP gene | Phase 1/2 | NCT05230459 | December 2028 |
| 2 | GNT0006 | Atamyo Therapeutics | GRT of FKRP gene | Phase 1/2 | NCT05224505 | October 2030 |
| Drug | Sponsor/Collaborators | Target of Gene Therapy | Status | NCT Number | Study Completion | |
|---|---|---|---|---|---|---|
| 1 | AOC 1001 | Avidity Biosciences | siRNA that targets DMPK mRNA, conjugated with a monoclonal antibody that binds to TfR1 | Phase 1/2 | NCT05027269 | September 2023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamakioti, M.; Karantzelis, N.; Taraviras, S. Advanced Gene-Targeting Therapies for Motor Neuron Diseases and Muscular Dystrophies. Int. J. Mol. Sci. 2022, 23, 4824. https://doi.org/10.3390/ijms23094824
Chamakioti M, Karantzelis N, Taraviras S. Advanced Gene-Targeting Therapies for Motor Neuron Diseases and Muscular Dystrophies. International Journal of Molecular Sciences. 2022; 23(9):4824. https://doi.org/10.3390/ijms23094824
Chicago/Turabian StyleChamakioti, Myrsini, Nikolaos Karantzelis, and Stavros Taraviras. 2022. "Advanced Gene-Targeting Therapies for Motor Neuron Diseases and Muscular Dystrophies" International Journal of Molecular Sciences 23, no. 9: 4824. https://doi.org/10.3390/ijms23094824
APA StyleChamakioti, M., Karantzelis, N., & Taraviras, S. (2022). Advanced Gene-Targeting Therapies for Motor Neuron Diseases and Muscular Dystrophies. International Journal of Molecular Sciences, 23(9), 4824. https://doi.org/10.3390/ijms23094824