
Targeting Cell Death Mechanism Specifically in Triple Negative Breast Cancer Cell Lines


 , ,
, ,  , ,
, ,  , ,
, ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
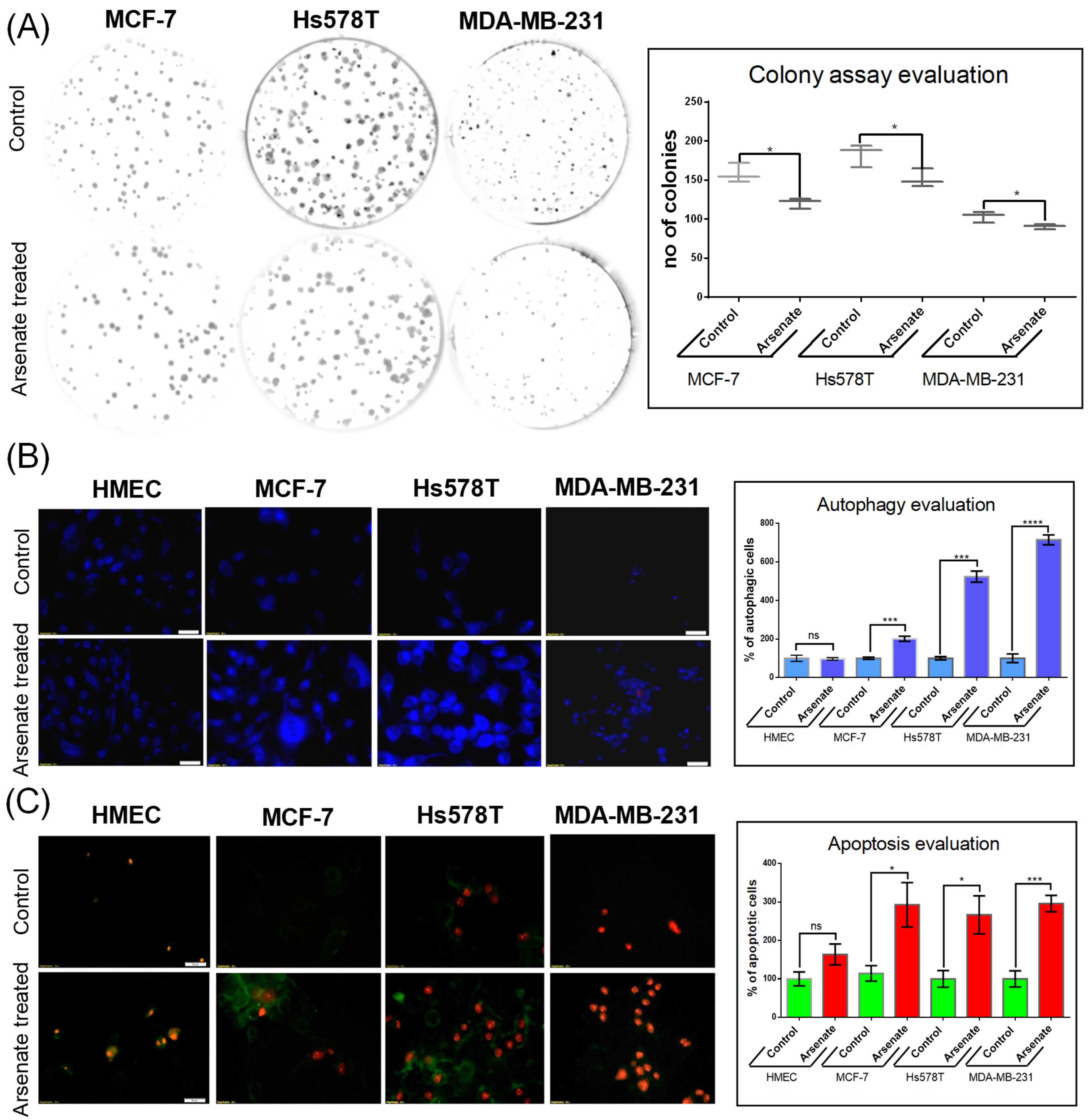
2.1. Arsenate Exposure Decreases Colony Formation in Cancer Cell Lines
2.2. Effect of Arsenate on the Modulation of Apoptosis and Autophagy in MCF-7, Hs578T, and MDA-MB-231 Cells
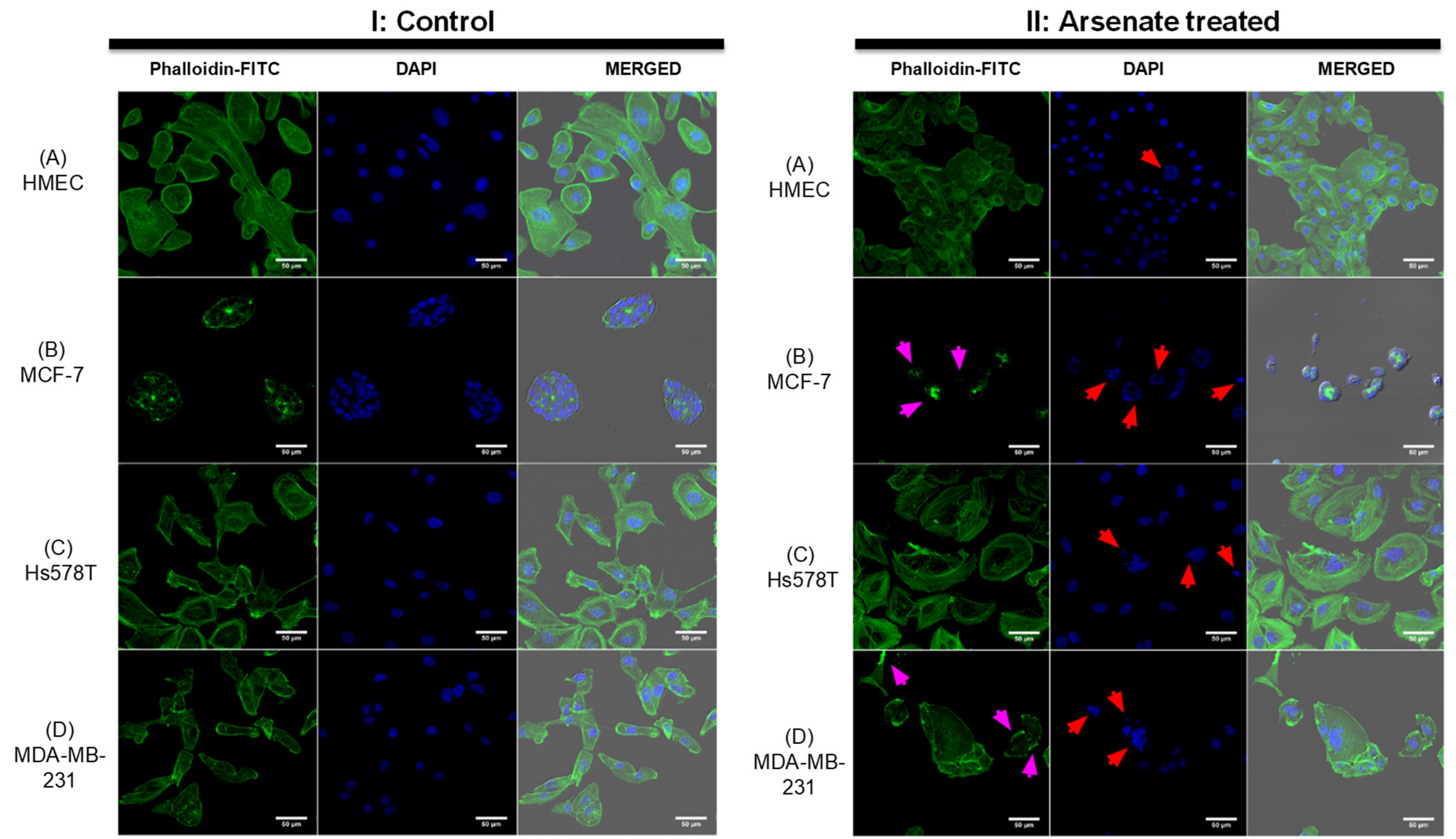
2.3. Dark-Field Microscopic and Cytoskeletal Evaluation
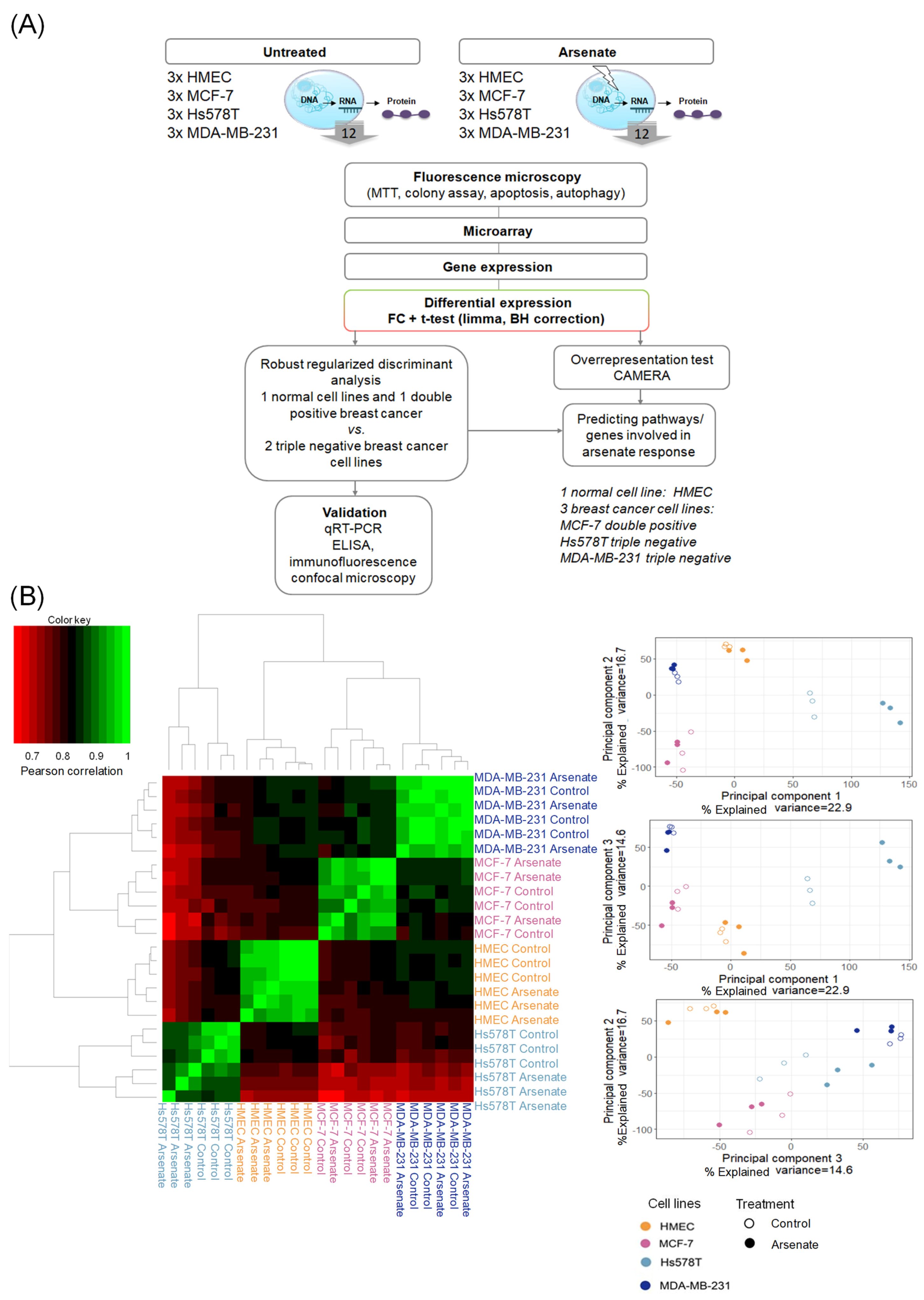
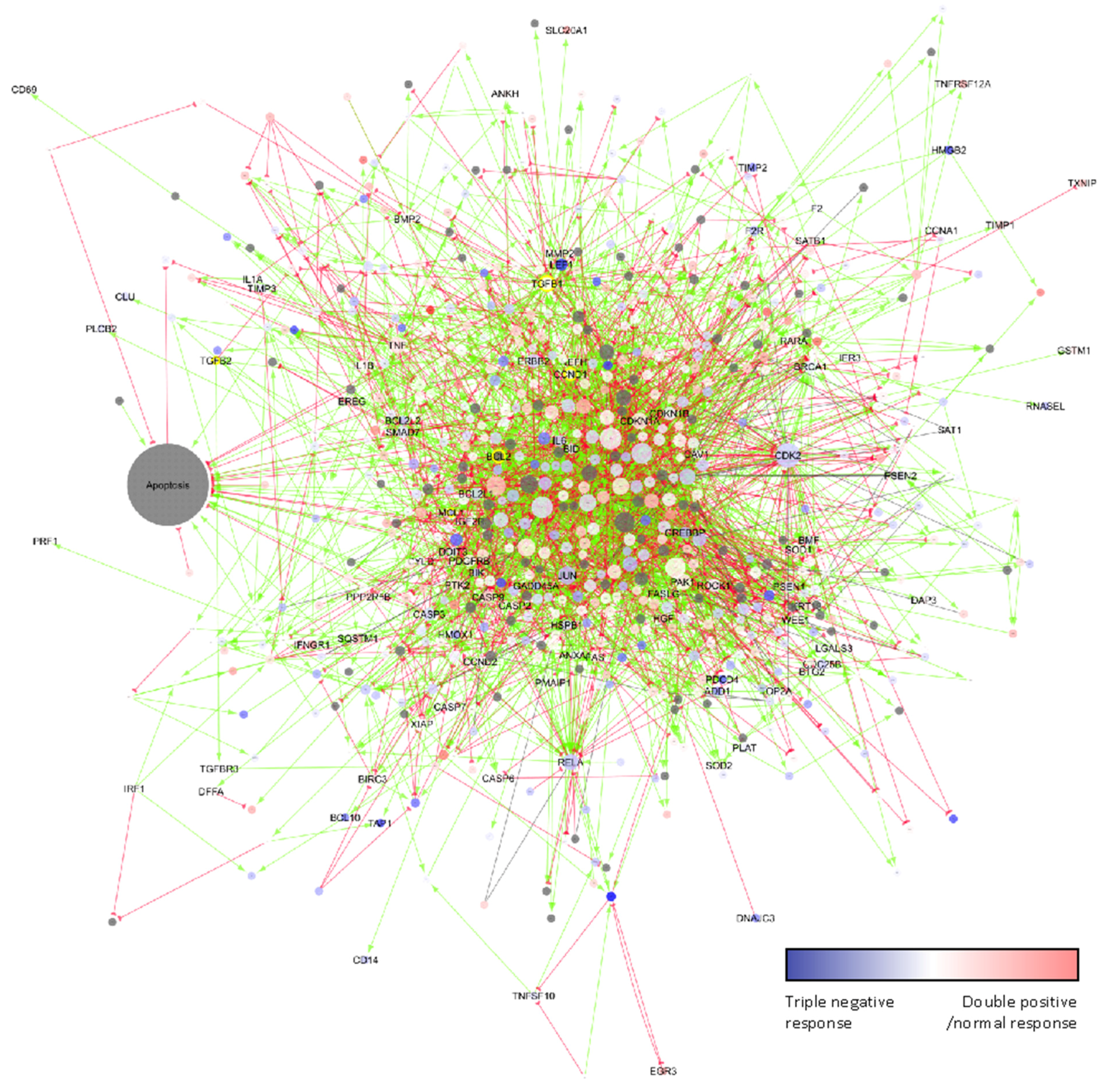
2.4. Mode-of-Action Analysis of Arsenate Treatment Based on Gene Expression Data
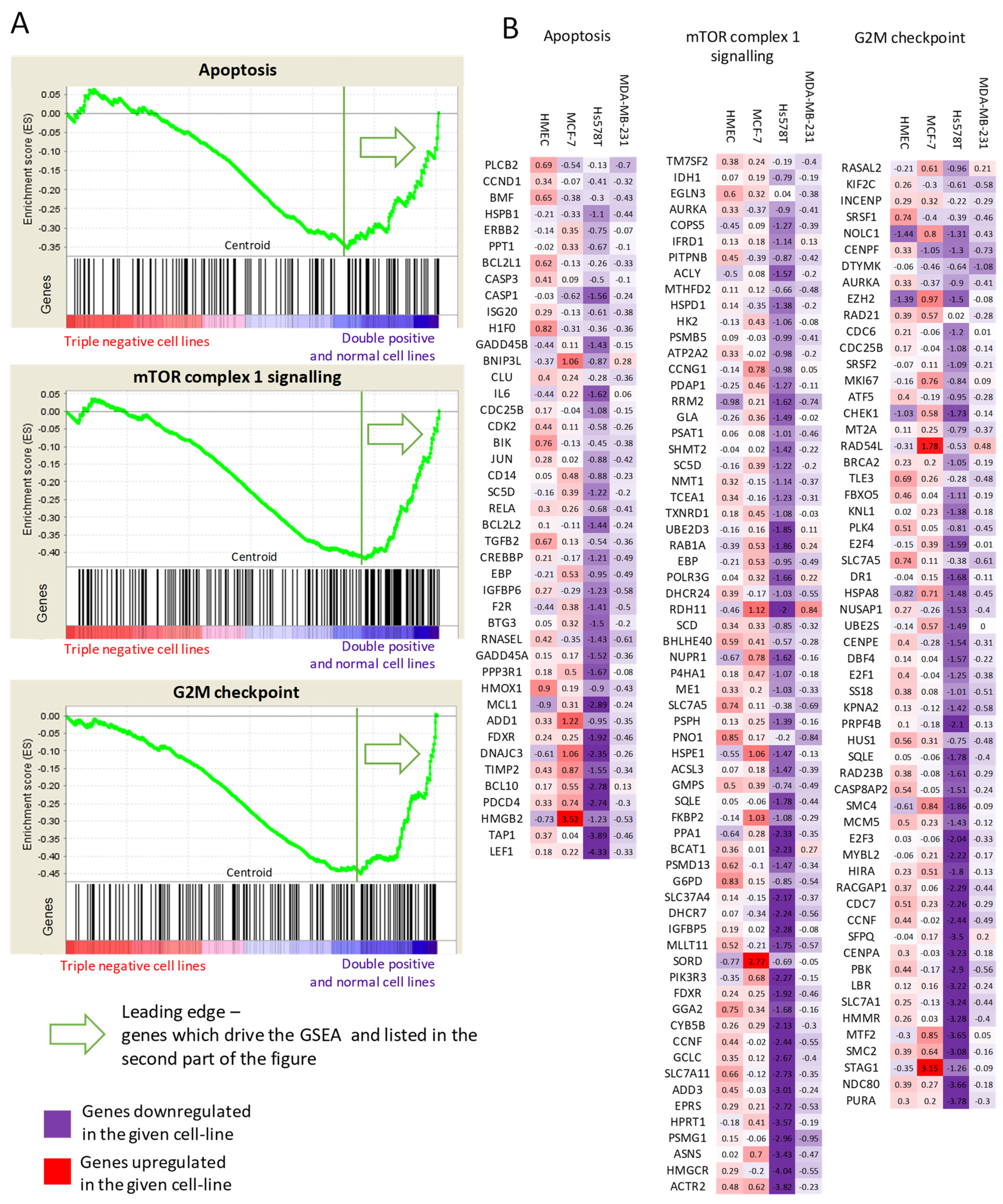
2.5. Arsenate Response in Triple Negative Cell Lines vs. Double Positive and Normal Cell Line
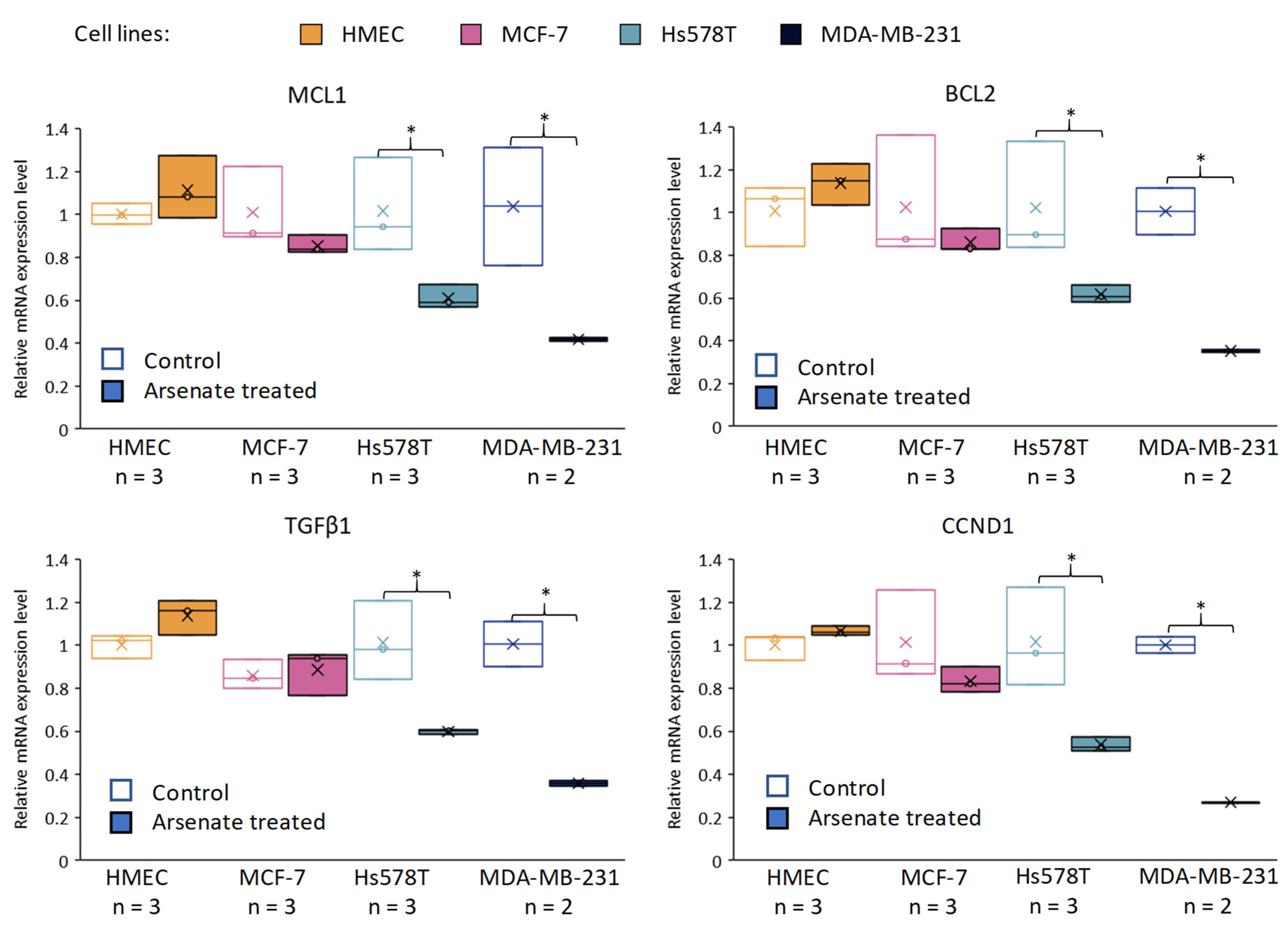
2.6. qRT-PCR Validation of Transcriptomic Profiles
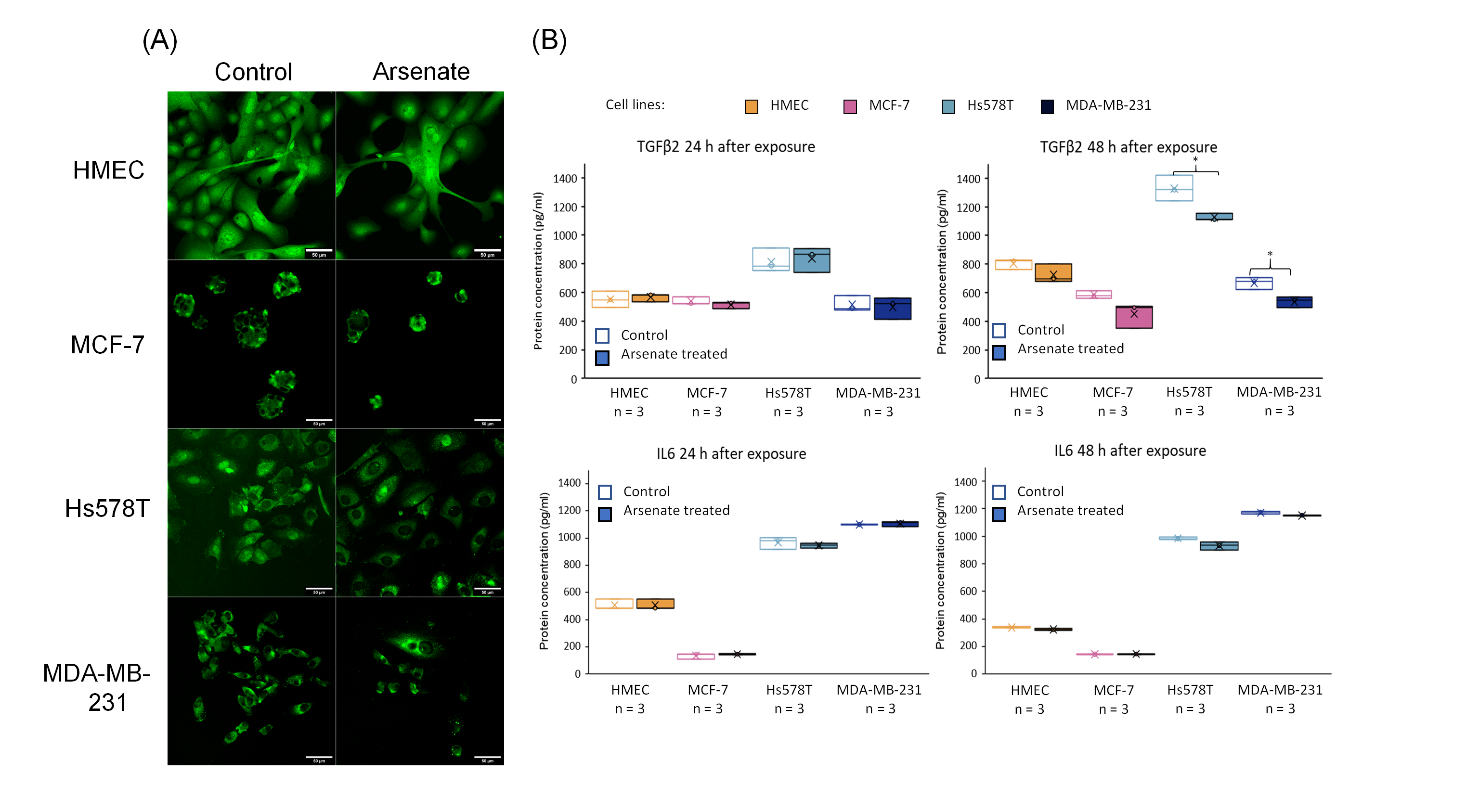
2.7. BCL2 Quantification by Fluorescence Confocal Microscopy and TGFβ2 Protein Quantification via ELISA
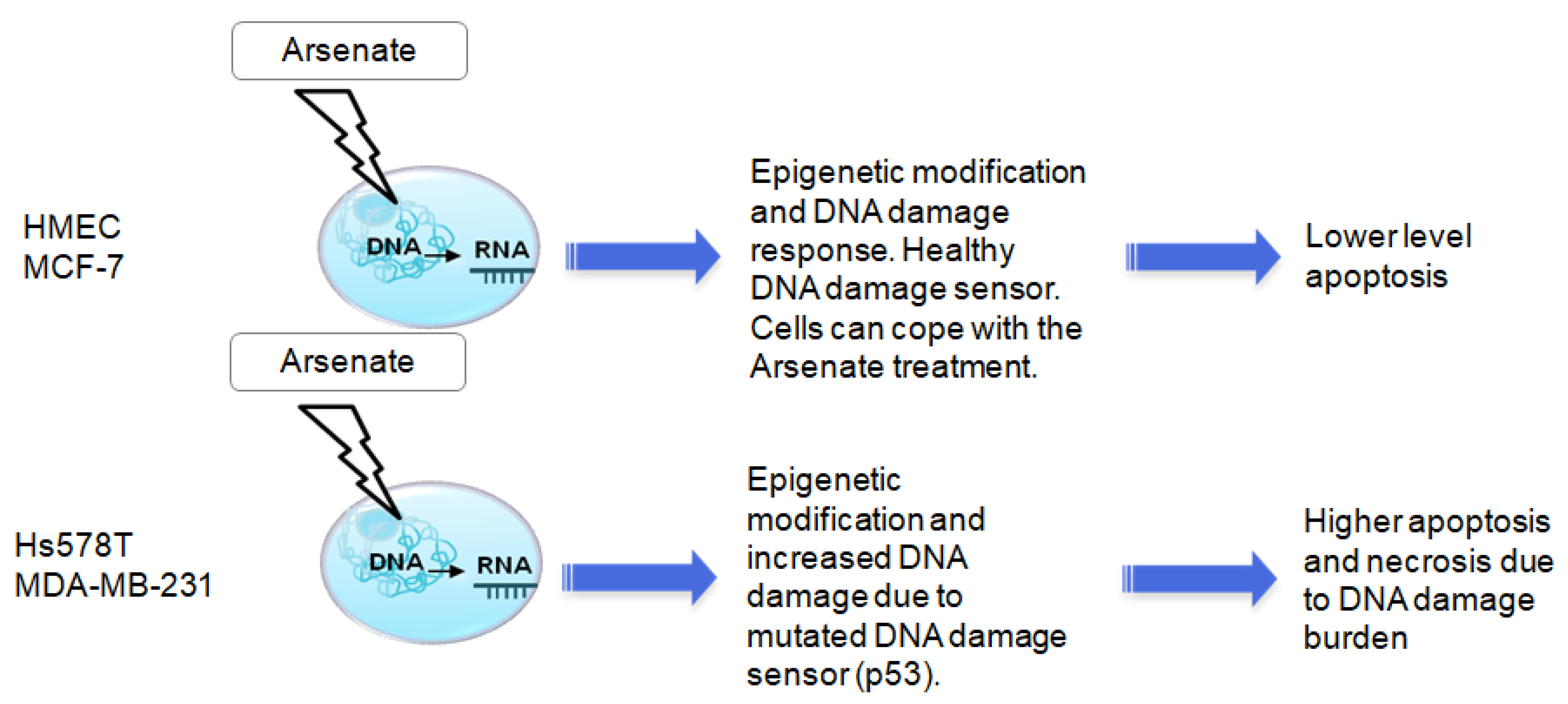
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Treatment
4.2. Colony Assay
4.3. Autophagy and Apoptosis Detection
4.4. Dark-Field Microscopy
4.5. Cytoskeletal Evaluation
4.6. Microarrays
4.7. Apoptosis Network in Pathological Condition as Effect of Arsenate Treatment
4.8. qRT-PCR Evaluation
4.9. TGFβ2 and IL6 Quantification in Cell Culture Medium
4.10. BCL2 Protein Evaluation by Confocal Microscopy
4.11. Statistical Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. IMpassion130 Trial Investigators Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, R.; Braicu, C.; Berindan-Neagoe, I. Another review on triple negative breast cancer. Are we on the right way towards the exit from the labyrinth? Breast 2013, 22, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Berindan-Neagoe, I.; Pileczki, V.; Cojocneanu-Petric, R.; Pop, L.-A.; Puscas, E.; Irimie, A.; Buiga, R. Breast tumor bank: An important resource for developing translational cancer research in Romania. Cancer Biomark. 2014, 14, 119–127. [Google Scholar] [CrossRef]
- Braicu, C.; Chiorean, R.; Irimie, A.; Chira, S.; Tomuleasa, C.; Neagoe, E.; Paradiso, A.; Achimas-Cadariu, P.; Lazar, V.; Berindan-Neagoe, I. Novel insight into triple-negative breast cancers, the emerging role of angiogenesis, and antiangiogenic therapy. Expert Rev. Mol. Med. 2016, 18, e18. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Yun, S.-M.; Woo, S.H.; Oh, S.T.; Hong, S.-E.; Choe, T.-B.; Ye, S.-K.; Kim, E.-K.; Seong, M.K.; Kim, H.-A.; Noh, W.C.; et al. Melatonin enhances arsenic trioxide-induced cell death via sustained upregulation of Redd1 expression in breast cancer cells. Mol. Cell. Endocrinol. 2016, 422, 64–73. [Google Scholar] [CrossRef]
- Moghaddaskho, F.; Eyvani, H.; Ghadami, M.; Tavakkoly-Bazzaz, J.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Demethylation and alterations in the expression level of the cell cycle-related genes as possible mechanisms in arsenic trioxide-induced cell cycle arrest in human breast cancer cells. Tumor Biol. 2017, 39, 1010428317692255. [Google Scholar] [CrossRef]
- Kasukabe, T.; Okabe-Kado, J.; Kato, N.; Honma, Y.; Kumakura, S. Cotylenin A and arsenic trioxide cooperatively suppress cell proliferation and cell invasion activity in human breast cancer cells. Int. J. Oncol. 2015, 46, 841–848. [Google Scholar] [CrossRef]
- Baj, G.; Arnulfo, A.; Deaglio, S.; Mallone, R.; Vigone, A.; De Cesaris, M.G.; Surico, N.; Malavasi, F.; Ferrero, E. Arsenic trioxide and breast cancer: Analysis of the apoptotic, differentiative and immunomodulatory effects. Breast Cancer Res. Treat. 2002, 73, 61–73. [Google Scholar] [CrossRef]
- Smith, A.H.; Marshall, G.; Yuan, Y.; Steinmaus, C.; Liaw, J.; Smith, M.T.; Wood, L.; Heirich, M.; Fritzemeier, R.M.; Pegram, M.D.; et al. Rapid reduction in breast cancer mortality with inorganic arsenic in drinking water. EBioMedicine 2014, 1, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Lacerda-Abreu, M.A.; Russo-Abrahão, T.; Monteiro, R. de Q.; Rumjanek, F.D.; Meyer-Fernandes, J.R. Inorganic phosphate transporters in cancer: Functions, molecular mechanisms and possible clinical applications. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 291–298. [Google Scholar] [CrossRef]
- Bolan, N.; Mahimairaja, S.; Kunhikrishnan, A.; Seshadri, B.; Thangarajan, R. Bioavailability and ecotoxicity of arsenic species in solution culture and soil system: Implications to remediation. Environ. Sci. Pollut. Res. Int. 2015, 22, 8866–8875. [Google Scholar] [CrossRef] [PubMed]
- Ratnaike, R.N. Acute and chronic arsenic toxicity. Postgrad. Med. J. 2003, 79, 391–396. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans Arsenic, metals, fibres, and dusts. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 11–465.
- Patterson, T.J.; Ngo, M.; Aronov, P.A.; Reznikova, T.V.; Green, P.G.; Rice, R.H. Biological activity of inorganic arsenic and antimony reflects oxidation state in cultured human keratinocytes. Chem. Res. Toxicol. 2003, 16, 1624–1631. [Google Scholar] [CrossRef]
- Carter, D.E.; Aposhian, H.V.; Gandolfi, A.J. The metabolism of inorganic arsenic oxides, gallium arsenide, and arsine: A toxicochemical review. Toxicol. Appl. Pharmacol. 2003, 193, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- Hartwig, A.; Blessing, H.; Schwerdtle, T.; Walter, I. Modulation of DNA repair processes by arsenic and selenium compounds. Toxicology 2003, 193, 161–169. [Google Scholar] [CrossRef]
- Russo-Abrahão, T.; Lacerda-Abreu, M.A.; Gomes, T.; Cosentino-Gomes, D.; Carvalho-de-Araújo, A.D.; Rodrigues, M.F.; de Oliveira, A.C.L.; Rumjanek, F.D.; de Monteiro, R.Q.; Meyer-Fernandes, J.R. Characterization of inorganic phosphate transport in the triple-negative breast cancer cell line, MDA-MB-. PLoS ONE 2018, 13, e0191270. [Google Scholar] [CrossRef]
- Li, Y.; Liu, K.-Q.; Gong, B.-F.; Wang, Y.; Wei, H.; Lin, D.; Liu, B.-C.; Zhou, C.-L.; Wei, S.-N.; Zhang, G.-J.; et al. Efficacy of Arsenic Trioxide Combined with ATRA and Chemotherapy for Relapsed Acute Promyelocytic Leukemia Patients. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 1–6. [Google Scholar] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, U.; Sommer, A.; Beckmann, G.; Lutzenberger, M.; Seidel, H.; Kreft, B.; Toschi, L. Isogenic human mammary epithelial cell lines: Novel tools for target identification and validation. Comprehensive characterization of an isogenic human mammary epithelial cell model provides evidence for epithelial-mesenchymal transition. Breast Cancer Res. Treat. 2013, 138, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Onda, K.; Sugiyama, K.; Yuan, B.; Tanaka, S.; Takagi, N.; Hirano, T. Antitumor effects of arsenic disulfide on the viability, migratory ability, apoptosis and autophagy of breast cancer cells. Oncol. Rep. 2019, 41, 27–42. [Google Scholar]
- Shi, Y.; Cao, T.; Huang, H.; Lian, C.; Yang, Y.; Wang, Z.; Ma, J.; Xia, J. Arsenic trioxide inhibits cell growth and motility via up-regulation of let-7a in breast cancer cells. Cell Cycle 2017, 16, 2396–2403. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, C.; Pang, H.; Zeng, F.; Cheng, L.; Fang, B.; Ma, J.; Shi, Y.; Hong, H.; Chen, J.; et al. Arsenic trioxide suppresses cell growth and migration via inhibition of miR-27a in breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 469, 55–61. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Yin, C.; An, B.; Hao, Y.; Wei, T.; Li, L.; Song, G. Arsenic trioxide inhibits breast cancer cell growth via microRNA-328/hERG pathway in MCF-7 cells. Mol. Med. Rep. 2015, 12, 1233–1238. [Google Scholar] [CrossRef][Green Version]
- Liu, W.; Gong, Y.; Li, H.; Jiang, G.; Zhan, S.; Liu, H.; Wu, Y. Arsenic trioxide-induced growth arrest of breast cancer MCF-7 cells involving FOXO3a and IκB kinase β expression and localization. Cancer Biother. Radiopharm. 2012, 27, 504–512. [Google Scholar] [CrossRef]
- Qi, Y.; Li, H.; Zhang, M.; Zhang, T.; Frank, J.; Chen, G. Autophagy in arsenic carcinogenesis. Exp. Toxicol. Pathol. 2014, 66, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ciocan-Cȃrtiţă, C.A.; Jurj, A.; Raduly, L.; Cojocneanu, R.; Moldovan, A.; Pileczki, V.; Pop, L.-A.; Budişan, L.; Braicu, C.; Korban, S.S.; et al. New perspectives in triple-negative breast cancer therapy based on treatments with TGFβ1 siRNA and doxorubicin. Mol. Cell. Biochem. 2020, 475, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Ciocan-Cartita, C.A.; Jurj, A.; Zanoaga, O.; Cojocneanu, R.; Pop, L.-A.; Moldovan, A.; Moldovan, C.; Zimta, A.A.; Raduly, L.; Pop-Bica, C.; et al. New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 241. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.E.; Dyck, L.; Laussmann, M.A.; Rehm, M. Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis. 2014, 5, e1011. [Google Scholar] [CrossRef]
- Zhao, Y.; Toselli, P.; Li, W. Microtubules as a critical target for arsenic toxicity in lung cells in vitro and in vivo. Int. J. Environ. Res. Public Health 2012, 9, 474–495. [Google Scholar] [CrossRef]
- Lencinas, A.; Broka, D.M.; Konieczka, J.H.; Klewer, S.E.; Antin, P.B.; Camenisch, T.D.; Runyan, R.B. Arsenic exposure perturbs epithelial-mesenchymal cell transition and gene expression in a collagen gel assay. Toxicol. Sci. 2010, 116, 273–285. [Google Scholar] [CrossRef]
- Troester, M.A.; Hoadley, K.A.; Sørlie, T.; Herbert, B.-S.; Børresen-Dale, A.-L.; Lønning, P.E.; Shay, J.W.; Kaufmann, W.K.; Perou, C.M. Cell-type-specific responses to chemotherapeutics in breast cancer. Cancer Res. 2004, 64, 4218–4226. [Google Scholar] [CrossRef]
- Bustaffa, E.; Stoccoro, A.; Bianchi, F.; Migliore, L. Genotoxic and epigenetic mechanisms in arsenic carcinogenicity. Arch. Toxicol. 2014, 88, 1043–1067. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Edlich, F. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; ten Dijke, P. TGF-β signaling in breast cancer cell invasion and bone metastasis. J. Mammary Gland Biol. Neoplasia 2011, 16, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Kozar, K.; Sicinski, P. Cell cycle progression without cyclin D-CDK4 and cyclin D-CDK6 complexes. Cell Cycle 2005, 4, 388–391. [Google Scholar] [CrossRef]
- Zagouri, F.; Kotoula, V.; Kouvatseas, G.; Sotiropoulou, M.; Koletsa, T.; Gavressea, T.; Valavanis, C.; Trihia, H.; Bobos, M.; Lazaridis, G.; et al. Protein expression patterns of cell cycle regulators in operable breast cancer. PLoS ONE 2017, 12, e0180489. [Google Scholar] [CrossRef]
- Lengare, P.V.; Sinai Khandeparkar, S.G.; Joshi, A.R.; Gogate, B.P.; Solanke, S.G.; Gore, S.H. Immunohistochemical expression of cyclin D1 in invasive breast carcinoma and its correlation with clinicopathological parameters. Indian J. Pathol Microbiol 2020, 63, 376–381. [Google Scholar]
- Tiainen, M.; Tammilehto, L.; Rautonen, J.; Tuomi, T.; Mattson, K.; Knuutila, S. Chromosomal abnormalities and their correlations with asbestos exposure and survival in patients with mesothelioma. Br. J. Cancer 1989, 60, 618–626. [Google Scholar] [CrossRef]
- Ahlin, C.; Lundgren, C.; Embretsén-Varro, E.; Jirström, K.; Blomqvist, C.; Fjällskog, M.L. High expression of cyclin D1 is associated to high proliferation rate and increased risk of mortality in women with ER-positive but not in ER-negative breast cancers. Breast Cancer Res. Treat. 2017, 164, 667–678. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Lan, S.-H.; Liu, H.-S. Degradative autophagy selectively regulates CCND1 (cyclin D1) and MIR224, two oncogenic factors involved in hepatocellular carcinoma tumorigenesis. Autophagy 2019, 15, 729–730. [Google Scholar] [CrossRef]
- Brown, N.E.; Jeselsohn, R.; Bihani, T.; Hu, M.G.; Foltopoulou, P.; Kuperwasser, C.; Hinds, P.W. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012, 72, 6477–6489. [Google Scholar] [CrossRef]
- Wang, X.; Gao, P.; Long, M.; Lin, F.; Wei, J.-X.; Ren, J.-H.; Yan, L.; He, T.; Han, Y.; Zhang, H.-Z. Essential role of cell cycle regulatory genes p21 and p27 expression in inhibition of breast cancer cells by arsenic trioxide. Med. Oncol. 2011, 28, 1225–1254. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Madani, S.; Gupta, I.; Shanmuganathan, S.; Abdraboh, M.E.; Al-Riyami, H.; Al-Farsi, Y.M.; Raj, M.H. TGF-β2: A Novel Target of CD44-Promoted Breast Cancer Invasion. J. Cancer 2013, 4, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Jung, J.; Moon, A.; Kang, H.; Cho, H. Antitumor and Anti-Invasive Effect of Apigenin on Human Breast Carcinoma through Suppression of IL-6 Expression. Int. J. Mol. Sci. 2019, 20, 3143. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Liu, K.; Gao, P.; Li, Z.; Zheng, J. Differential anticancer activities of arsenic trioxide on head and neck cancer cells with different human papillomavirus status. Life Sci. 2018, 212, 182–193. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, X.; Du, R.; Gao, W.; Luo, N.; Zhao, S.; Li, Y.; Chen, R.; Wang, H.; Bao, Y.; et al. Low dosage of arsenic trioxide (As2O3) inhibits angiogenesis in epithelial ovarian cancer without cell apoptosis. J. Biol. Inorg. Chem. 2018, 23, 939–947. [Google Scholar] [CrossRef]
- Xiong, X.; Li, Y.; Liu, L.; Qi, K.; Zhang, C.; Chen, Y.; Fang, J. Arsenic trioxide induces cell cycle arrest and affects Trk receptor expression in human neuroblastoma SK-N-SH cells. Biol. Res. 2018, 51, 18. [Google Scholar] [CrossRef]
- Sadaf, N.; Kumar, N.; Ali, M.; Ali, V.; Bimal, S.; Haque, R. Arsenic trioxide induces apoptosis and inhibits the growth of human liver cancer cells. Life Sci. 2018, 205, 9–17. [Google Scholar] [CrossRef]
- Mohammadi Kian, M.; Mohammadi, S.; Tavallaei, M.; Chahardouli, B.; Rostami, S.; Zahedpanah, M.; Ghavamzadeh, A.; Nikbakht, M. Inhibitory effects of arsenic trioxide and thalidomide on angiogenesis and vascular endothelial growth factor expression in leukemia cells. Asian Pac. J. Cancer Prev. 2018, 19, 1127–1134. [Google Scholar]
- Li, Y.-L.; Jin, Y.-F.; Liu, X.-X.; Li, H.-J. A comprehensive analysis of Wnt/β-catenin signaling pathway-related genes and crosstalk pathways in the treatment of As2O3 in renal cancer. Ren. Fail. 2018, 40, 331–339. [Google Scholar] [CrossRef]
- Ji, H.; Li, Y.; Jiang, F.; Wang, X.; Zhang, J.; Shen, J.; Yang, X. Inhibition of transforming growth factor beta/SMAD signal by MiR-155 is involved in arsenic trioxide-induced anti-angiogenesis in prostate cancer. Cancer Sci. 2014, 105, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Hiwatashi, Y.; Tadokoro, H.; Henmi, K.; Arai, M.; Kaise, T.; Tanaka, S.; Hirano, T. Antiproliferative and anti-invasive effects of inorganic and organic arsenic compounds on human and murine melanoma cells in vitro. J. Pharm. Pharmacol. 2011, 63, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Arai, M.; Tanaka, S.; Onda, K.; Sugiyama, K.; Hirano, T. Effects of arsenic compounds on growth, cell-cycle distribution and apoptosis of tretinoin-resistant human promyelocytic leukemia cells. Anticancer Res. 2014, 34, 6489–6494. [Google Scholar] [PubMed]
- Hikita, E.; Arai, M.; Tanaka, S.; Onda, K.; Utsumi, H.; Yuan, B.; Toyoda, H.; Hirano, T. Effects of inorganic and organic arsenic compounds on growth and apoptosis of human T-lymphoblastoid leukemia cells. Anticancer Res. 2011, 31, 4169–4178. [Google Scholar]
- Nagappan, A.; Lee, W.S.; Yun, J.W.; Lu, J.N.; Chang, S.-H.; Jeong, J.-H.; Kim, G.S.; Jung, J.-M.; Hong, S.C. Tetraarsenic hexoxide induces G2/M arrest, apoptosis, and autophagy via PI3K/Akt suppression and p38 MAPK activation in SW620 human colon cancer cells. PLoS ONE 2017, 12, e0174591. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramos, R.; Lopez-Carrillo, L.; Rios-Perez, A.D.; De Vizcaya-Ruíz, A.; Cebrian, M.E. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-kappaB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat. Res. 2009, 674, 109–115. [Google Scholar] [CrossRef]
- Zhao, Y.; Onda, K.; Yuan, B.; Tanaka, S.; Kiyomi, A.; Sugiyama, K.; Sugiura, M.; Takagi, N.; Hirano, T. Arsenic disulfide-induced apoptosis and its potential mechanism in two- and three-dimensionally cultured human breast cancer MCF-7 cells. Int. J. Oncol. 2018, 52, 1959–1971. [Google Scholar] [CrossRef]
- Wallace, D.R.; Taalab, Y.M.; Heinze, S.; Tariba Lovaković, B.; Pizent, A.; Renieri, E.; Tsatsakis, A.; Farooqi, A.A.; Javorac, D.; Andjelkovic, M.; et al. Toxic-Metal-Induced Alteration in miRNA Expression Profile as a Proposed Mechanism for Disease Development. Cells 2020, 9, 901. [Google Scholar] [CrossRef]
- Ghaffari, S.H.; Bashash, D.; Dizaji, M.Z.; Ghavamzadeh, A.; Alimoghaddam, K. Alteration in miRNA gene expression pattern in acute promyelocytic leukemia cell induced by arsenic trioxide: A possible mechanism to explain arsenic multi-target action. Tumor Biol. 2012, 33, 157–172. [Google Scholar] [CrossRef]
- Gao, J.; Wang, G.; Wu, J.; Zuo, Y.; Zhang, J.; Jin, X. Skp2 Expression Is Inhibited by Arsenic Trioxide through the Upregulation of miRNA-330-5p in Pancreatic Cancer Cells. Mol. Ther. Oncolytics 2019, 12, 214–223. [Google Scholar] [CrossRef]
- Wu, B.; Tan, M.; Cai, W.; Wang, B.; He, P.; Zhang, X. Arsenic trioxide induces autophagic cell death in osteosarcoma cells via the ROS-TFEB signaling pathway. Biochem. Biophys. Res. Commun. 2018, 496, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.L.; Lam, S.-K.; Li, Y.-Y.; Ho, J.C.-M. Tumour growth-suppressive effect of arsenic trioxide in squamous cell lung carcinoma. Oncol. Lett. 2017, 14, 3748–3754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pileczki, V.; Braicu, C.; Gherman, C.D.; Berindan-Neagoe, I. TNF-α gene knockout in triple negative breast cancer cell line induces apoptosis. Int. J. Mol. Sci. 2012, 14, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-Y.; Gong, L.-Y.; Liao, R.; Weng, N.-N.; Feng, Y.-Y.; Dong, Y.-P.; Zhu, H.; Zhao, Y.-Q.; Zhang, Y.-Y.; Zhu, Q.; et al. Evaluation of the target genes of arsenic trioxide in pancreatic cancer by bioinformatics analysis. Oncol. Lett. 2019, 18, 5163–5172. [Google Scholar] [CrossRef]
- Zhang, H.-W.; Hu, J.-J.; Fu, R.-Q.; Liu, X.; Zhang, Y.-H.; Li, J.; Liu, L.; Li, Y.-N.; Deng, Q.; Luo, Q.-S.; et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kγ mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci. Rep. 2018, 8, 11255. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Stevens, J.J.; Graham, B.; Dugo, E.; Berhaneselassie-Sumner, B.; Ndebele, K.; Tchounwou, P.B. Arsenic Trioxide Induces Apoptosis via Specific Signaling Pathways in HT-29 Colon Cancer Cells. J. Cancer Sci. Ther. 2017, 9, 298–306. [Google Scholar] [CrossRef]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between apoptosis and autophagy: Molecular mechanisms and therapeutic strategies in cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [CrossRef]
- Kharroubi, W.; Nury, T.; Ahmed, S.H.; Andreoletti, P.; Sakly, R.; Hammami, M.; Lizard, G. Induction by arsenate of cell-type-specific cytotoxic effects in nerve and hepatoma cells. Hum. Exp. Toxicol. 2017, 36, 1256–1269. [Google Scholar]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.A.; Fry, R.C. Arsenic-Associated Changes to the Epigenome: What Are the Functional Consequences? Curr. Environ. Health Rep. 2014, 1, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.G.; Gamble, M.V. Influence of arsenic on global levels of histone posttranslational modifications: A review of the literature and challenges in the field. Curr. Environ. Health Rep. 2016, 3, 225–237. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.M. Chromatin modifications during repair of environmental exposure-induced DNA damage: A potential mechanism for stable epigenetic alterations. Env. Mol. Mutagen. 2014, 55, 278–291. [Google Scholar] [CrossRef]
- Chaidez, B.R.; Dixon, K. Arsenic Exposure Alters a Chromatin Silencing Pathway. Epidemiology 2008, 19, S226. [Google Scholar] [CrossRef]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef]
- Mu, Y.-F.; Chen, Y.-H.; Chang, M.-M.; Chen, Y.-C.; Huang, B.-M. Arsenic compounds induce apoptosis through caspase pathway activation in MA-10 Leydig tumor cells. Oncol. Lett. 2019, 18, 944–954. [Google Scholar] [CrossRef]
- Treas, J.N.; Tyagi, T.; Singh, K.P. Effects of chronic exposure to arsenic and estrogen on epigenetic regulatory genes expression and epigenetic code in human prostate epithelial cells. PLoS ONE 2012, 7, e43880. [Google Scholar] [CrossRef]
- Muenyi, C.S.; Ljungman, M.; States, J.C. Arsenic Disruption of DNA Damage Responses-Potential Role in Carcinogenesis and Chemotherapy. Biomolecules 2015, 5, 2184–2193. [Google Scholar] [CrossRef]
- Nishikawa, S.; Ishii, H.; Haraguchi, N.; Kano, Y.; Fukusumi, T.; Ohta, K.; Ozaki, M.; Sakai, D.; Satoh, T.; Nagano, H.; et al. Genotoxic therapy stimulates error-prone DNA repair in dormant hepatocellular cancer stem cells. Exp. Med. 2012, 3, 959–962. [Google Scholar] [CrossRef][Green Version]
- Charoensuk, V.; Gati, W.P.; Weinfeld, M.; Le, X.C. Differential cytotoxic effects of arsenic compounds in human acute promyelocytic leukemia cells. Toxicol. Appl. Pharmacol. 2009, 239, 64–70. [Google Scholar] [CrossRef]
- Ventura-Lima, J.; Bogo, M.R.; Monserrat, J.M. Arsenic toxicity in mammals and aquatic animals: A comparative biochemical approach. Ecotoxicol. Environ. Saf. 2011, 74, 211–218. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 2017, 18, 529. [Google Scholar] [CrossRef]
- Braicu, C.; Cojocneanu-Petric, R.; Jurj, A.; Gulei, D.; Taranu, I.; Gras, A.M.; Marin, D.E.; Berindan-Neagoe, I. Microarray based gene expression analysis of Sus Scrofa duodenum exposed to zearalenone: Significance to human health. BMC Genom. 2016, 17, 646. [Google Scholar] [CrossRef]
- Petric, R.C.; Braicu, C.; Bassi, C.; Pop, L.; Taranu, I.; Dragos, N.; Dumitrascu, D.; Negrini, M.; Berindan-Neagoe, I. Interspecies Gene Name Extrapolation--A New Approach. PLoS ONE 2015, 10, e0138751. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Core Team, R. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2015. [Google Scholar]
- Pruteanu, L.-L.; Kopanitsa, L.; Módos, D.; Kletnieks, E.; Samarova, E.; Bender, A.; Gomez, L.D.; Bailey, D.S. Transcriptomics predicts compound synergy in drug and natural product treated glioblastoma cells. PLoS ONE 2020, 15, e0239551. [Google Scholar] [CrossRef]
- Phipson, B.; Lee, S.; Majewski, I.J.; Alexander, W.S.; Smyth, G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef]
- Guo, Y.; Hastie, T.; Tibshirani, R. Regularized linear discriminant analysis and its application in microarrays. Biostatistics 2007, 8, 86–100. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- UniProt Consortium UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [CrossRef]
- Perfetto, L.; Briganti, L.; Calderone, A.; Cerquone Perpetuini, A.; Iannuccelli, M.; Langone, F.; Licata, L.; Marinkovic, M.; Mattioni, A.; Pavlidou, T.; et al. SIGNOR: A database of causal relationships between biological entities. Nucleic Acids Res. 2016, 44, D548–D554. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pruteanu, L.-L.; Braicu, C.; Módos, D.; Jurj, M.-A.; Raduly, L.-Z.; Zănoagă, O.; Magdo, L.; Cojocneanu, R.; Paşca, S.; Moldovan, C.; et al. Targeting Cell Death Mechanism Specifically in Triple Negative Breast Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 4784. https://doi.org/10.3390/ijms23094784
Pruteanu L-L, Braicu C, Módos D, Jurj M-A, Raduly L-Z, Zănoagă O, Magdo L, Cojocneanu R, Paşca S, Moldovan C, et al. Targeting Cell Death Mechanism Specifically in Triple Negative Breast Cancer Cell Lines. International Journal of Molecular Sciences. 2022; 23(9):4784. https://doi.org/10.3390/ijms23094784
Chicago/Turabian StylePruteanu, Lavinia-Lorena, Cornelia Braicu, Dezső Módos, Maria-Ancuţa Jurj, Lajos-Zsolt Raduly, Oana Zănoagă, Lorand Magdo, Roxana Cojocneanu, Sergiu Paşca, Cristian Moldovan, and et al. 2022. "Targeting Cell Death Mechanism Specifically in Triple Negative Breast Cancer Cell Lines" International Journal of Molecular Sciences 23, no. 9: 4784. https://doi.org/10.3390/ijms23094784
APA StylePruteanu, L.-L., Braicu, C., Módos, D., Jurj, M.-A., Raduly, L.-Z., Zănoagă, O., Magdo, L., Cojocneanu, R., Paşca, S., Moldovan, C., Moldovan, A. I., Ţigu, A. B., Gurzău, E., Jäntschi, L., Bender, A., & Berindan-Neagoe, I. (2022). Targeting Cell Death Mechanism Specifically in Triple Negative Breast Cancer Cell Lines. International Journal of Molecular Sciences, 23(9), 4784. https://doi.org/10.3390/ijms23094784