
Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Fish, Reagents and Antibodies
2.2. Chemical Treatment and Phenotype Observation
2.3. RNA-seq
2.4. Quantitative PCR
2.5. Whole-Mount in Situ Hybridization (WISH)
2.6. Intracellular ROS and Redox Enzymes Detection
2.7. Transmission Electron Microscopy (TEM)
2.8. Cryosection
2.9. Immunofluorescence and Apoptosis Assays
2.10. FerroOrange Staining
2.11. Statistical Analysis
3. Results
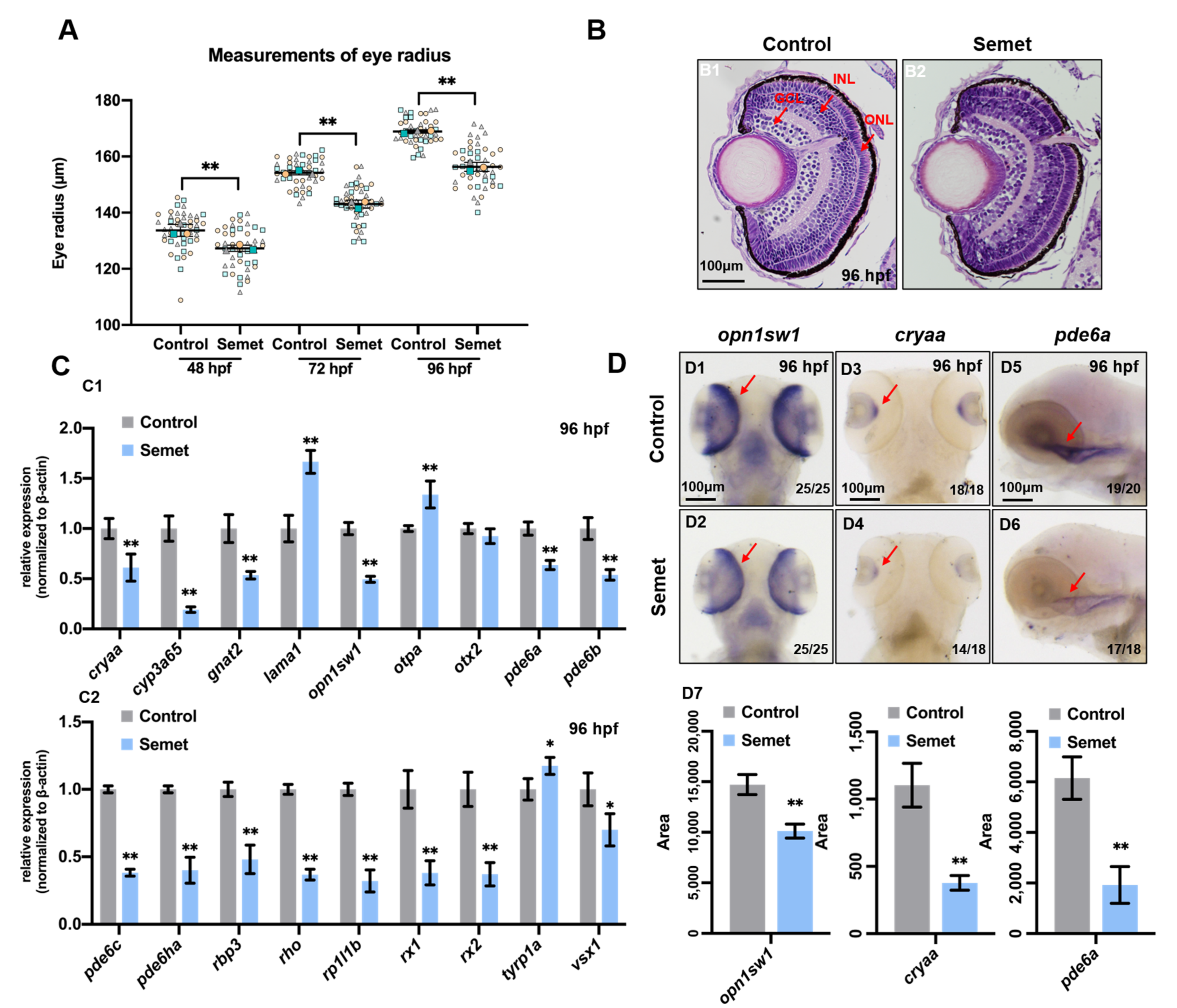
3.1. Excessive Selenium-Induced Ocular Defects in Zebrafish Embryos
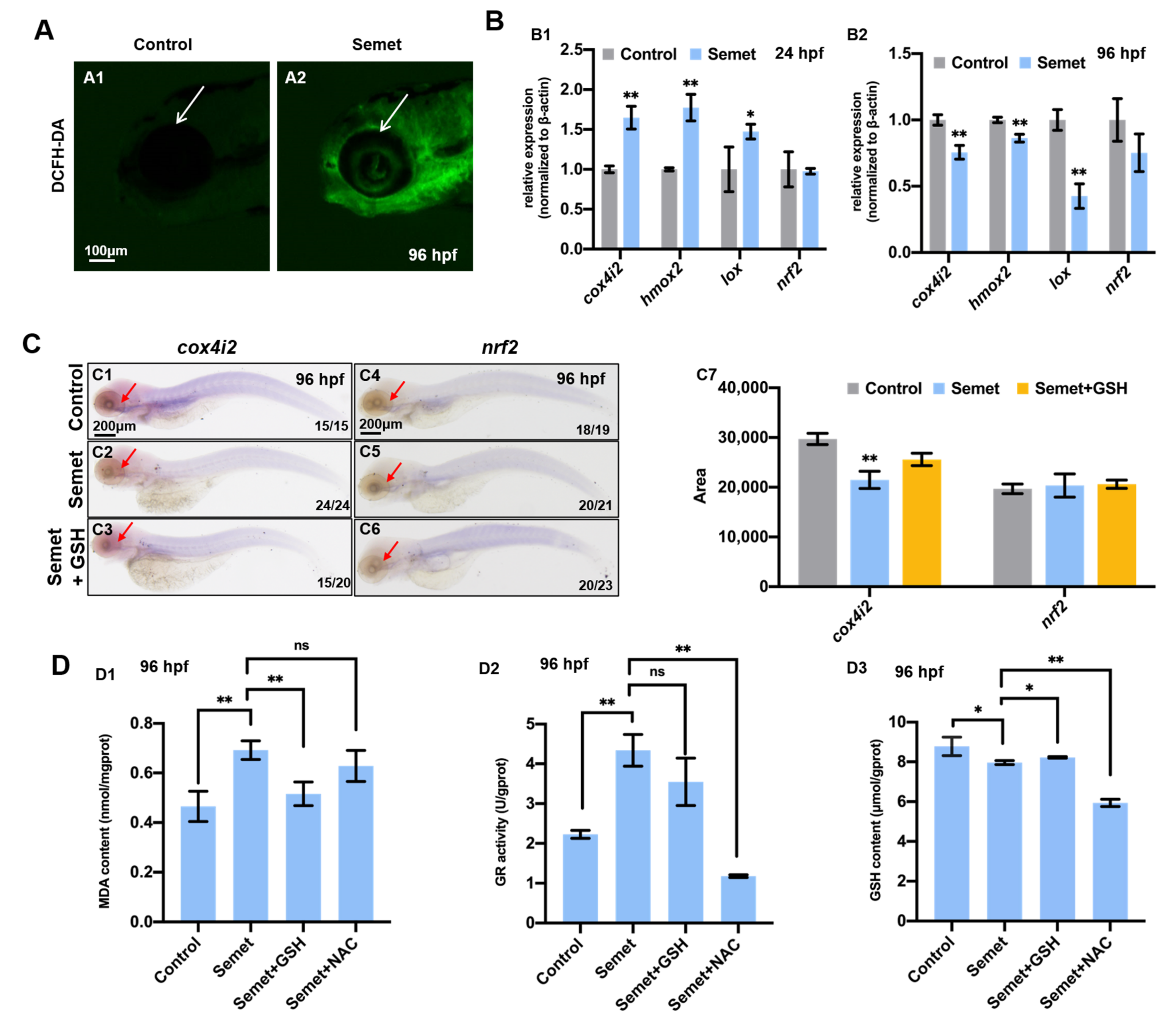
3.2. Excessive Selenium Caused ROS Accumulation and Oxidative Stress in Embryonic Cells
3.3. Excessive Selenium Triggered Cell Apoptosis and Proliferation in Zebrafish Eyes
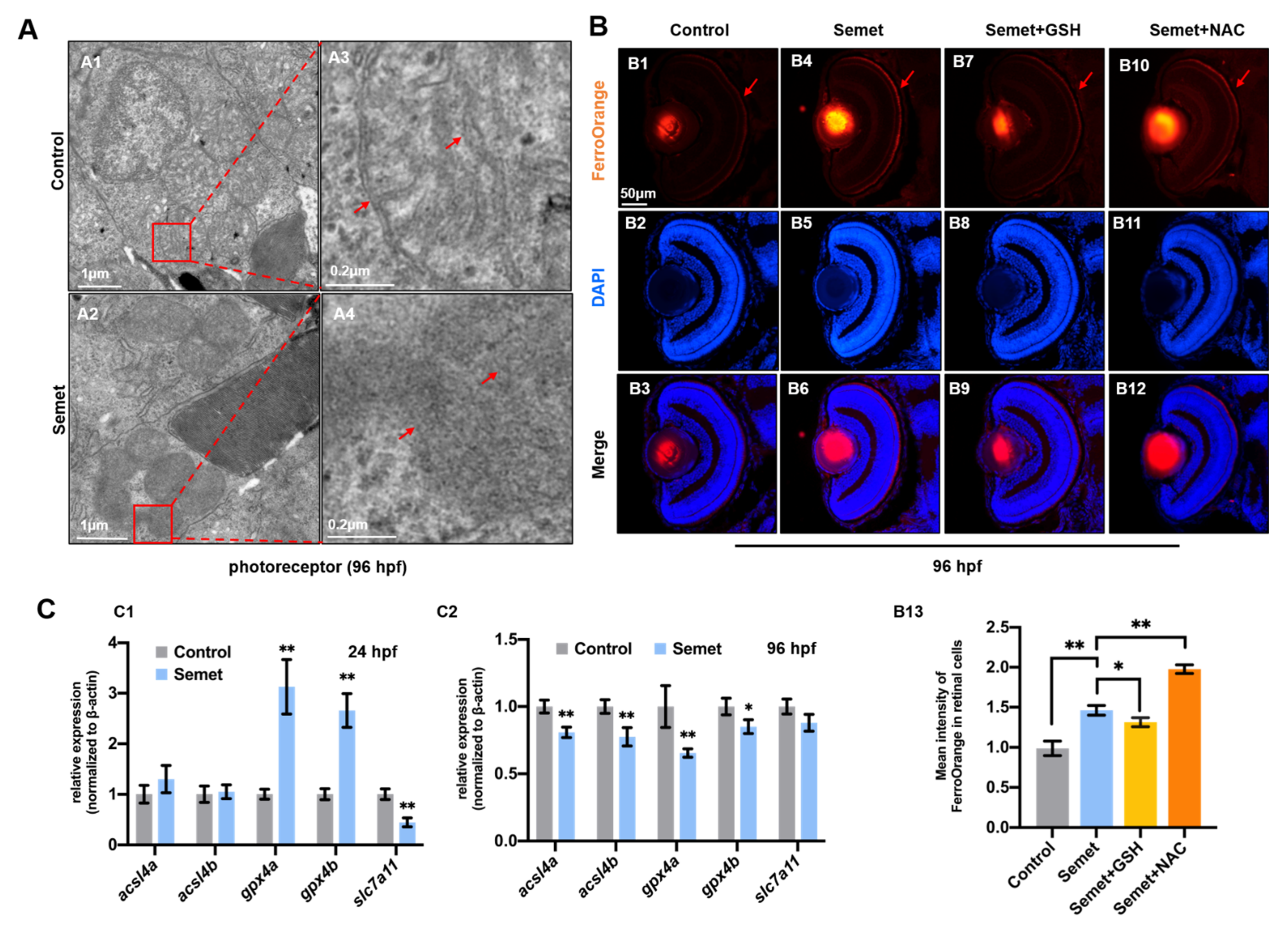
3.4. Excessive Selenium-Induced Ferroptosis in Eye Cells
3.5. Scavenging of ROS Failed to Rescue Selenium-Induced Eye Defects
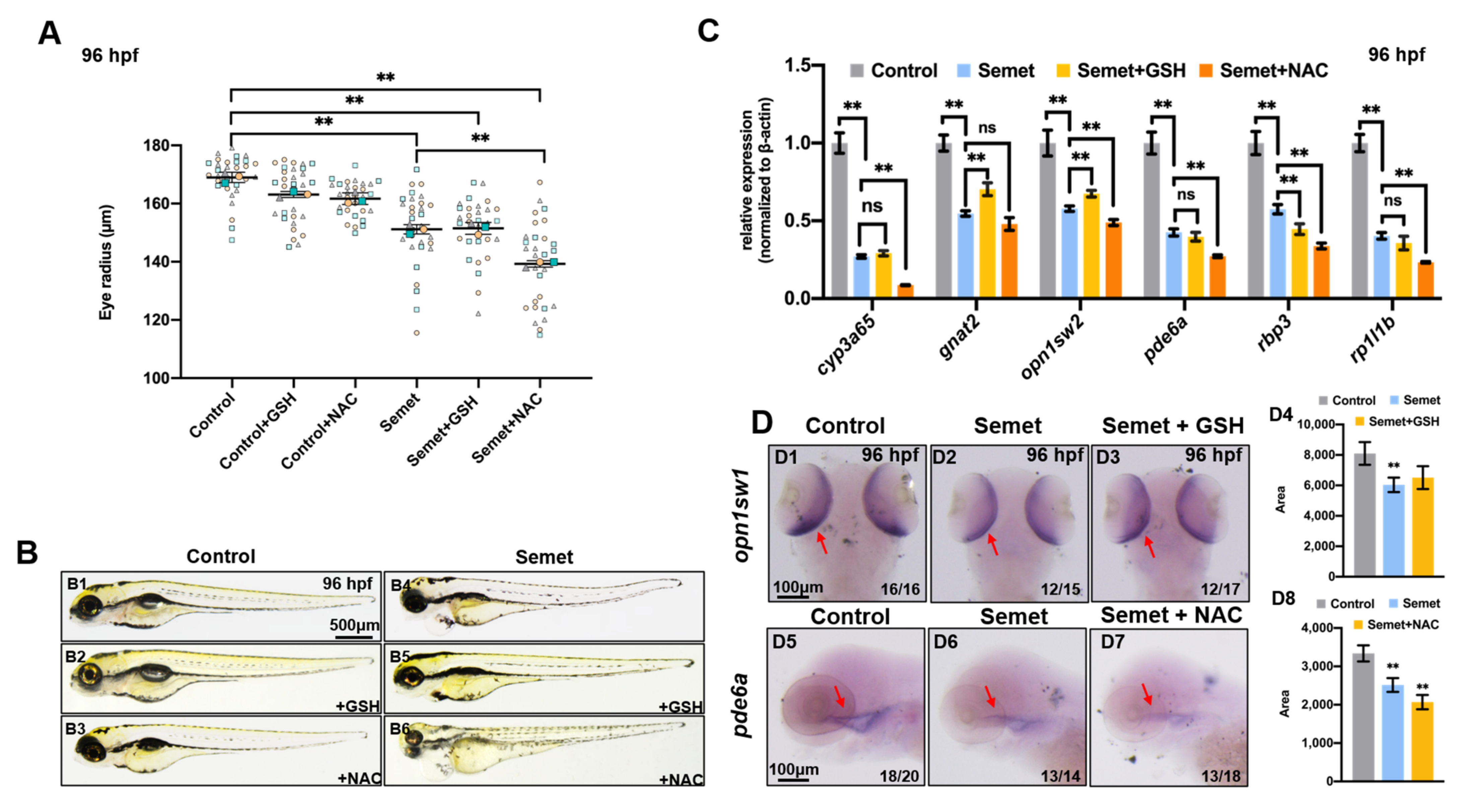
3.6. Promotion of Ferroptosis and Apoptosis Rescued the Eye Defects in Selenium-Stressed Embryos
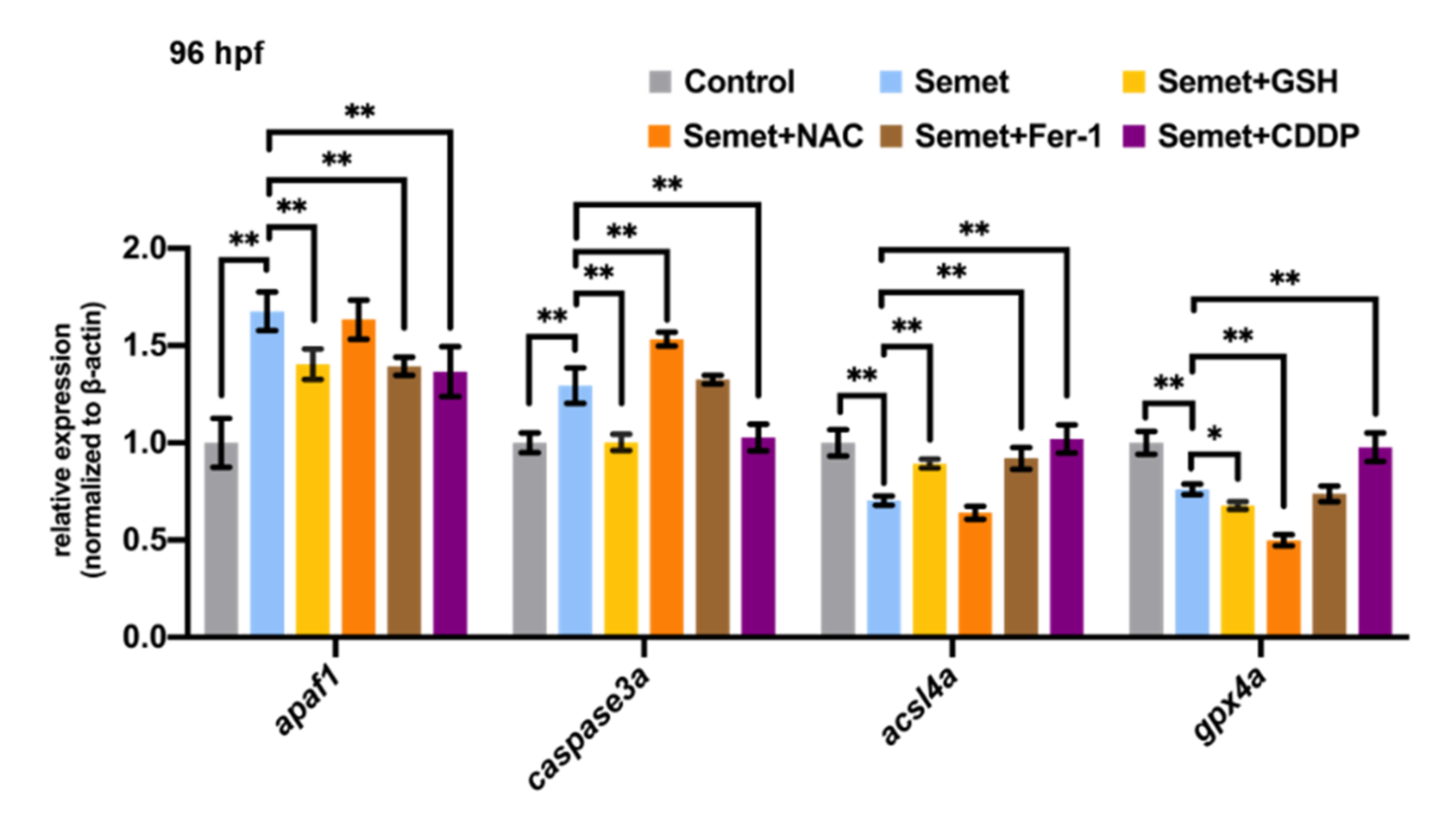
3.7. NAC, GSH, Fer-1 and CDDP Altered the Expression of Genes Related to Apoptosis and Ferroptosis
4. Discussion
4.1. Toxic Effect of Excessive Selenium on Zebrafish Eyes
4.2. Effects of Excessive Selenium on Apoptosis, Proliferation and Ferroptosis of Embryonic Eye Cells
4.3. Effects of NAC, GSH, Fer-1 and CDDP on Selenium-Induced Zebrafish Embryonic Microphthalmia Defects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Thomas, J.K.; Wiseman, S.; Giesy, J.P.; Janz, D.M. Effects of chronic dietary selenomethionine exposure on repeat swimming performance, aerobic metabolism and methionine catabolism in adult zebrafish (Danio rerio). Aquat. Toxicol. 2013, 130, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Selenius, M.; Rundlöf, A.-K.; Olm, E.; Fernandes, A.P.; Björnstedt, M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox Signal. 2010, 12, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Özkaya, D.; Nazıroğlu, M.; Vanyorek, L.; Muhamad, S. Involvement of TRPM2 Channel on Hypoxia-Induced Oxidative Injury, Inflammation, and Cell Death in Retinal Pigment Epithelial Cells: Modulator Action of Selenium Nanoparticles. Biol. Trace Elem. Res. 2021, 199, 1356–1369. [Google Scholar] [CrossRef] [PubMed]
- Ananth, S.; Miyauchi, S.; Thangaraju, M.; Jadeja, R.N.; Bartoli, M.; Ganapathy, V.; Martin, P.M. Selenomethionine (Se-Met) induces the cystine/glutamate exchanger SLC7A11 in cultured human retinal pigment epithelial (rpe) cells: Implications for antioxidant therapy in aging retina. Antioxidants 2021, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Kohler, L.N.; Foote, J.; Kelley, C.P.; Florea, A.; Shelly, C.; Chow, H.; Hsu, P.; Batai, K.; Ellis, N.; Saboda, K. Selenium and type 2 diabetes: Systematic review. Nutrients 2018, 10, 1924. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.M.; Darke, A.K.; Penney, K.L.; Tangen, C.M.; Goodman, P.J.; Lee, G.-S.M.; Sun, T.; Peisch, S.; Tinianow, A.M.; Rae, J.M. Selenium-or vitamin E–related gene variants, interaction with supplementation, and risk of high-grade prostate cancer in SELECT. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Vinceti, M.; Bonvicini, F.; Rothman, K.J.; Vescovi, L.; Wang, F. The relation between amyotrophic lateral sclerosis and inorganic selenium in drinking water: A population-based case-control study. Environ. Health 2010, 9, 77. [Google Scholar] [CrossRef] [Green Version]
- Ellwanger, J.H.; Franke, S.I.; Bordin, D.L.; Pra, D.; Henriques, J.A. Biological functions of selenium and its potential influence on Parkinson’s disease. An. Acad. Bras. Cienc. 2016, 88, 1655–1674. [Google Scholar] [CrossRef] [Green Version]
- Shini, S.; Sultan, A.; Bryden, W.L. Selenium biochemistry and bioavailability: Implications for animal agriculture. Agriculture 2015, 5, 1277–1288. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.W.-M.; Teh, S.J.; Hinton, D.E.; Higashi, R.M. Selenium biotransformations into proteinaceous forms by foodweb organisms of selenium-laden drainage waters in California. Aquat. Toxicol. 2002, 57, 65–84. [Google Scholar] [CrossRef]
- Etim, E.U. Occurrence and distribution of arsenic, antimony and selenium in shallow groundwater systems of Ibadan Metropolis, Southwestern Nigerian. J. Health Pollut. 2017, 7, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Luo, K.; Du, Y.; Tian, Y.; Long, J.; Zhao, X.; Zhang, S. Hydrochemical characteristics of natural water and selenium-rich water resources in the Northern Daba Mountains, China. J. Water Health 2017, 15, 273–287. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, Y.; Bai, J.; Liang, N.; Wang, C.; Wang, P. Study on the Content of Selenium in Fish Muscles and the Bioaccumulation Effect in Bosten Lake, Xinjiang. Northwest. Geol. 2021, 54, 236–243. [Google Scholar]
- Chen, L. Visual system: An understudied target of aquatic toxicology. Aquat. Toxicol. 2020, 225, 105542. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, M.; Thomas, P.A.; Teresa, P.A.; Anbukkarasi, M.; Geraldine, P. Regulatory effect of chrysin on expression of lenticular calcium transporters, calpains, and apoptotic-cascade components in selenite-induced cataract. Mol. Vis. 2016, 22, 401. [Google Scholar]
- Valavanidis, A.; Vlahogianni, T.; Dassenakis, M.; Scoullos, M. Molecular biomarkers of oxidative stress in aquatic organisms in relation to toxic environmental pollutants. Ecotoxicol. Environ. Saf. 2006, 64, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Misra, S.; Wasik, A.M.; Marzano, C.; Björnstedt, M.; Gandin, V.; Fernandes, A.P. Selenium induces a multi-targeted cell death process in addition to ROS formation. J. Cell. Mol. Med. 2014, 18, 671–684. [Google Scholar] [CrossRef]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCORD, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive Oxygen Species (ROS)––Induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res.-Fundam. Mol. Mech. Mutag. 2011, 711, 167–173. [Google Scholar] [CrossRef]
- Liu, H.; Gooneratne, R.; Huang, X.; Lai, R.; Wei, J.; Wang, W. A rapid in vivo zebrafish model to elucidate oxidative stress-mediated PCB126-induced apoptosis and developmental toxicity. Free Radic. Biol. Med. 2015, 84, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Mugoni, V.; Camporeale, A.; Santoro, M.M. Analysis of oxidative stress in zebrafish embryos. J. Vis. Exp. 2014, 89, e51328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Kimura, M.; Itokawa, Y.; Aoki, T.; Takahashi, J.A.; Nakatsu, S.; Oda, Y.; Kikuchi, H. Apoptosis induced by selenium in human glioma cell lines. Biol. Trace Elem. Res. 1996, 54, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Tomblin, J.; Armistead, M.Y.; Murray, E. Selenium (sodium selenite) causes cytotoxicity and apoptotic mediated cell death in PLHC-1 fish cell line through DNA and mitochondrial membrane potential damage. Ecotoxicol. Environ. Saf. 2013, 87, 80–88. [Google Scholar] [CrossRef]
- Ung, L.; Pattamatta, U.; Carnt, N.; Wilkinson-Berka, J.L.; Liew, G.; White, A.J. Oxidative stress and reactive oxygen species: A review of their role in ocular disease. Clin. Sci. 2017, 131, 2865–2883. [Google Scholar] [CrossRef]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.; Ma, J.-X. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H738–H749. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, J.K.; Ruemmler, R.; Hartmann, E.K.; Ziebart, A.; Ludwig, M.; Patzak, A.; Xia, N.; Li, H.; Pfeiffer, N.; Gericke, A. Responses of retinal arterioles and ciliary arteries in pigs with acute respiratory distress syndrome (ARDS). Exp. Eye Res. 2019, 184, 152–161. [Google Scholar] [CrossRef]
- Li, L.; Fan, D.-B.; Zhao, Y.-T.; Li, Y.; Yang, Z.-B.; Zheng, G.-Y. GJA8 missense mutation disrupts hemichannels and induces cell apoptosis in human lens epithelial cells. Sci. Rep. 2019, 9, 19157. [Google Scholar] [CrossRef]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef]
- Schmitt, E.A.; Dowling, J.E. Early-eye morphogenesis in the zebrafish, Brachydanio rerio. J. Comp. Neurol. 1994, 344, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.A.; Dowling, J.E. Early retinal development in the zebrafish, Danio rerio: Light and electron microscopic analyses. J. Comp. Neurol. 1999, 404, 515–536. [Google Scholar] [CrossRef]
- Zhao, G.; Zhu, Y.; Hu, J.; Gao, M.; Hong, Y. l-selenomethionine induces zebrafish embryo cardiovascular defects via down-regulating expression of lrp2b. Chemosphere 2021, 64, 133351. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-X.; Hu, B.; Wang, Y.; Gui, J.-F.; Xiao, W. Zebrafish eaf1 and eaf2/u19 mediate effective convergence and extension movements through the maintenance of wnt11 and wnt5 expression. J. Biol. Chem. 2009, 284, 16679–16692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, S.J.; Velle, K.B.; Mullins, R.D.; Fritz-Laylin, L.K. SuperPlots: Communicating reproducibility and variability in cell biology. J. Cell Biol. 2020, 219, e202001064. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.; Ungureanu, G.; Boaventura, R.; Botelho, C. Selenium contaminated waters: An overview of analytical methods, treatment options and recent advances in sorption methods. Sci. Total Environ. 2015, 521, 246–260. [Google Scholar] [CrossRef]
- Attaran, A.; Salahinejad, A.; Crane, A.L.; Niyogi, S.; Chivers, D.P. Chronic exposure to dietary selenomethionine dysregulates the genes involved in serotonergic neurotransmission and alters social and antipredator behaviours in zebrafish (Danio rerio). Environ. Pollut. 2019, 246, 837–844. [Google Scholar] [CrossRef]
- Mo, A.; Wang, X.; Yuan, Y.; Liu, C.; Wang, J. Effects of waterborne exposure to environmentally relevant concentrations of selenite on reproductive function of female zebrafish: A life cycle assessment. Environ. Pollut. 2021, 270, 116237. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, M.; Li, D.; Li, X.-Q.; Li, P.; Zhao, J.; Luo, M.-N.; Guo, C.-L.; Gao, X.-B.; Lu, C.-L. Embryonic developmental toxicity of selenite in zebrafish (Danio rerio) and prevention with folic acid. Food Chem. Toxicol. 2012, 50, 2854–2863. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human gene mutation database (HGMD®): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Si, N.; Xiao, W. A novel mutation in the CRYAA gene associated with congenital cataract and microphthalmia in a Chinese family. BMC Med. Genet. 2018, 19, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, R.H. Characteristics of photoreceptor PDE (PDE6): Similarities and differences to PDE5. Int. J. Impot. Res. 2004, 16, S28–S33. [Google Scholar] [CrossRef] [Green Version]
- Zang, J.; Neuhauss, S.C. Biochemistry and physiology of zebrafish photoreceptors. Pflug. Arch. 2021, 473, 1569–1585. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Corbett, C.; Klaska, I.P.; Makinen, K.; Nickerson, J.M.; Cornall, R.J.; Kuffova, L.; Forrester, J.V. Partial retinal photoreceptor loss in a transgenic mouse model associated with reduced levels of interphotoreceptor retinol binding protein (IRBP, RBP3). Exp. Eye Res. 2018, 172, 54–65. [Google Scholar] [CrossRef]
- Noel, N.C.; Nadolski, N.J.; Hocking, J.C.; MacDonald, I.M.; Allison, W.T. Progressive photoreceptor dysfunction and age-related macular degeneration-like features in rp1l1 mutant zebrafish. Cells 2020, 9, 2214. [Google Scholar] [CrossRef]
- Tang, P.H.; Fan, J.; Goletz, P.W.; Wheless, L.; Crouch, R.K. Effective and sustained delivery of hydrophobic retinoids to photoreceptors. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5958–5964. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, J.; Geng, H.; Li, L.; Li, J.; Cheng, B.; Ma, X.; Li, H.; Hou, L. Photoreceptor degeneration in microphthalmia (Mitf) mice: Partial rescue by pigment epithelium-derived factor. Dis. Model. Mech. 2019, 12, dmm035642. [Google Scholar] [CrossRef] [Green Version]
- Al Jothery, A.H.; Vaanholt, L.M.; Mody, N.; Arnous, A.; Lykkesfeldt, J.; Bünger, L.; Hill, W.G.; Mitchell, S.E.; Allison, D.B.; Speakman, J.R. Oxidative costs of reproduction in mouse strains selected for different levels of food intake and which differ in reproductive performance. Sci. Rep. 2016, 6, 36353. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, D. Contaminant-stimulated reactive oxygen species production and oxidative damage in aquatic organisms. Mar. Pollut. Bull. 2001, 42, 656–666. [Google Scholar] [CrossRef]
- Frank, R.N.; Amin, R.H.; Puklin, J.E. Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. Am. J. Ophthalmol. 1999, 127, 694–709. [Google Scholar] [CrossRef]
- Lucero, H.; Kagan, H. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Revelles, S.; García-Redondo, A.B.; Avendaño, M.S.; Varona, S.; Palao, T.; Orriols, M.; Roque, F.R.; Fortuño, A.; Touyz, R.M.; Martínez-González, J. Lysyl oxidase induces vascular oxidative stress and contributes to arterial stiffness and abnormal elastin structure in hypertension: Role of p38MAPK. Antioxid. Redox Signal. 2017, 27, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Cykowiak, M.; Szaefer, H.; Kleszcz, R.; Baer-Dubowska, W. Combination of xanthohumol and phenethyl isothiocyanate inhibits NF-κB and activates Nrf2 in pancreatic cancer cells. Toxicol. In Vitro 2020, 65, 104799. [Google Scholar] [CrossRef]
- Trueba, G.P.; Sánchez, G.M.; Giuliani, A. Oxygen free radical and antioxidant defense mechanism in cancer. Front. Biosci. 2004, 9, 2029–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gönenç, A.; Hacışevki, A.; Aslan, S.; Torun, M.; Şimşek, B. Increased oxidative DNA damage and impaired antioxidant defense system in patients with gastrointestinal cancer. Eur. J. Intern. Med. 2012, 23, 350–354. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Rajavel, T.; Packiyaraj, P.; Suryanarayanan, V.; Singh, S.K.; Ruckmani, K.; Devi, K.P. β-Sitosterol targets Trx/Trx1 reductase to induce apoptosis in A549 cells via ROS mediated mitochondrial dysregulation and p53 activation. Sci. Rep. 2018, 8, 2071. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Huang, L.; He, J.; Cai, S.; Weng, Y.; Huang, S.; Ma, S. PTEN inhibits non-small cell lung cancer cell growth by promoting G0/G1 arrest and cell apoptosis. Oncol. Lett. 2019, 17, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Kong, Y.-Y.; Yoshida, R.; Elia, A.J.; Hakem, A.; Hakem, R.; Penninger, J.M.; Mak, T.W. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998, 94, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Yu, T.; Zhang, Q.; Zhang, H.; Xiao, M.; Cui, S.; Zhao, Y.; Lu, X. Malignant transformation of human bronchial epithelial cells induced by benzo [a] pyrene suggests a negative feedback of TP53 to PPP1R13L via binding a possible enhancer element. Chem. Biol. Interact. 2021, 349, 109683. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, R.; Xu, Y.; Yuan, C.; Sheng, X.; Wu, Z.; Peng, J.; Wang, Y.; Lin, Y.; Zhou, L.; Xu, S. Predictive and prognostic impact of ferroptosis-related genes ACSL4 and GPX4 on breast cancer treated with neoadjuvant chemotherapy. EBioMedicine 2021, 71, 103560. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, X.; Cheng, Y.; Yang, M.; Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 2020, 34, 16262–16275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox. Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Loehrer, P.J.; Einhorn, L.H. Cisplatin. Ann. Intern. Med. 1984, 100, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Yazici, Z.M.; Meric, A.; Midi, A.; Arınc, Y.V.; Kahya, V.; Hafız, G. Reduction of cisplatin ototoxicity in rats by oral administration of pomegranate extract. Eur. Arch. Otorhinolaryngol. 2012, 269, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Gregg, R.W.; Molepo, J.M.; Monpetit, V.; Mikael, N.Z.; Redmond, D.; Gadia, M.; Stewart, D.J. Cisplatin neurotoxicity: The relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J. Clin. Oncol. 1992, 10, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Yazici, A.; Sogutlu-Sari, E.; Yay, A.; Aksit, H.; Kilic, A.; Aksit, D.; Yildiz, O.; Ermis, S.S. The protective effect of selenium in cisplatin-related retinotoxicity. Cutan. Ocul. Toxicol. 2014, 33, 327–332. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Hu, J.; Zhu, Y.; Wang, X.; Zeng, S.; Hong, Y.; Zhao, G. Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos. Int. J. Mol. Sci. 2022, 23, 4783. https://doi.org/10.3390/ijms23094783
Gao M, Hu J, Zhu Y, Wang X, Zeng S, Hong Y, Zhao G. Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos. International Journal of Molecular Sciences. 2022; 23(9):4783. https://doi.org/10.3390/ijms23094783
Chicago/Turabian StyleGao, Meng, Jun Hu, Yuejie Zhu, Xianqing Wang, Shumin Zeng, Yijiang Hong, and Guang Zhao. 2022. "Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos" International Journal of Molecular Sciences 23, no. 9: 4783. https://doi.org/10.3390/ijms23094783
APA StyleGao, M., Hu, J., Zhu, Y., Wang, X., Zeng, S., Hong, Y., & Zhao, G. (2022). Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos. International Journal of Molecular Sciences, 23(9), 4783. https://doi.org/10.3390/ijms23094783