
Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway


 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
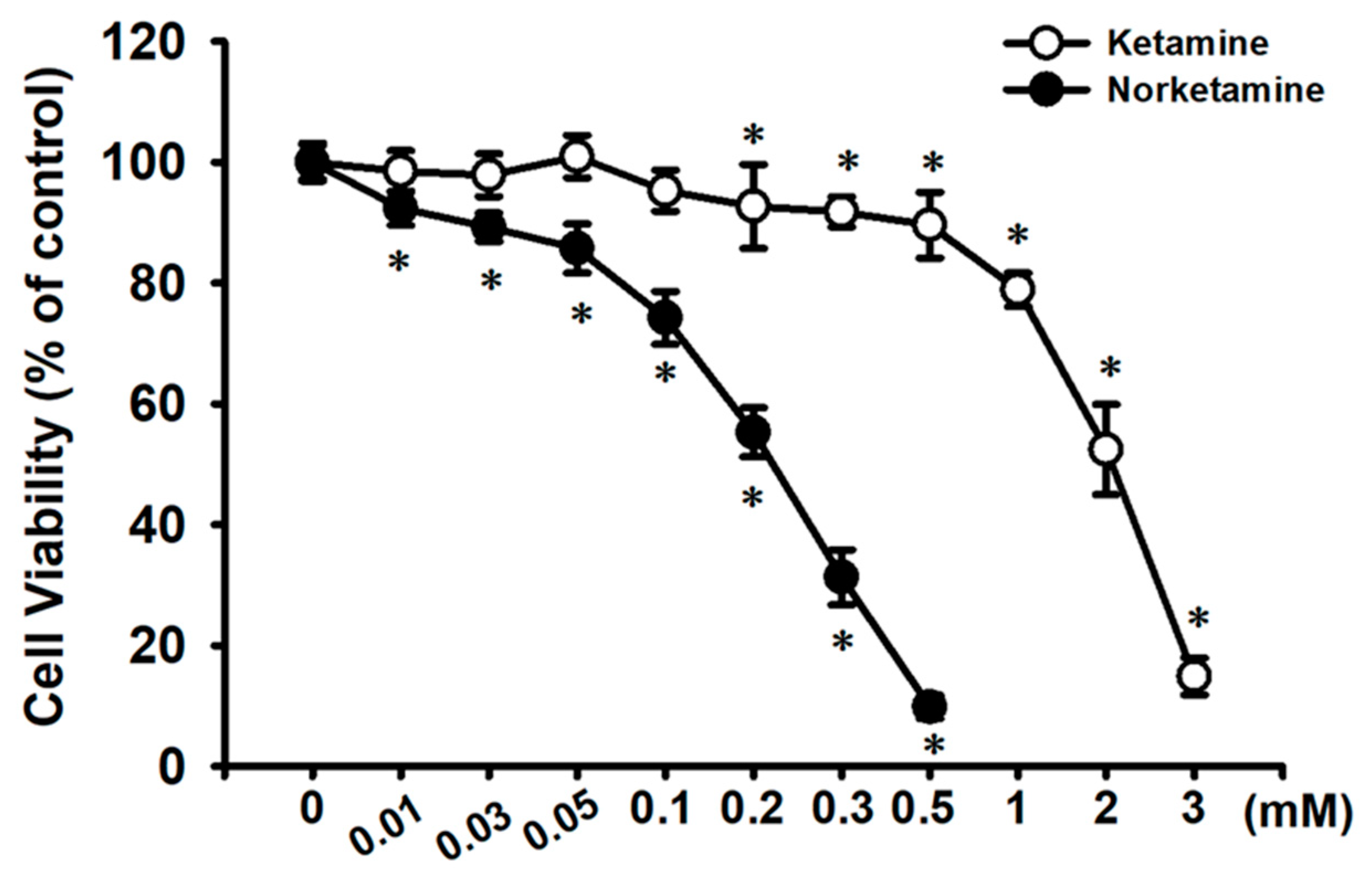
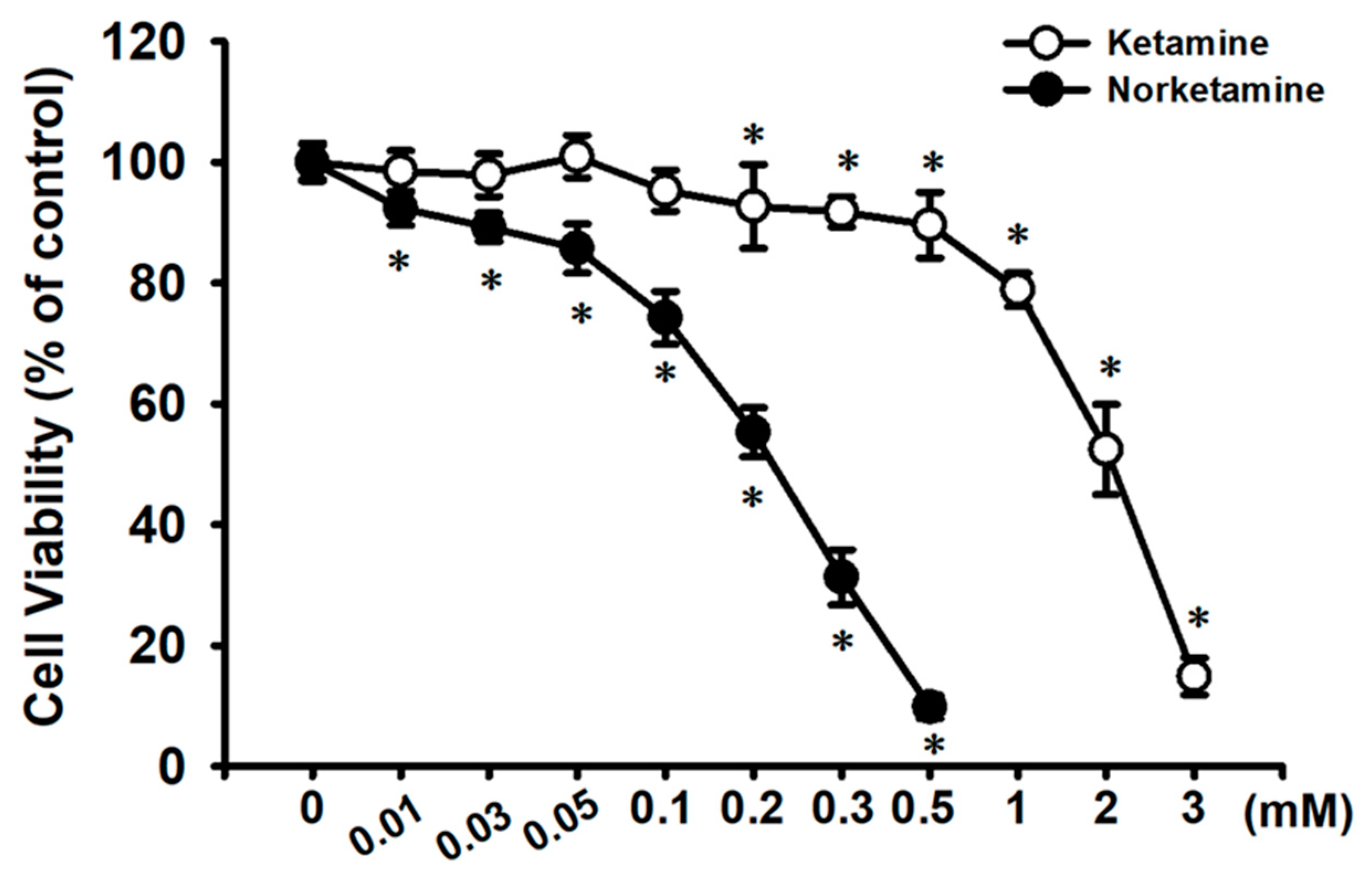
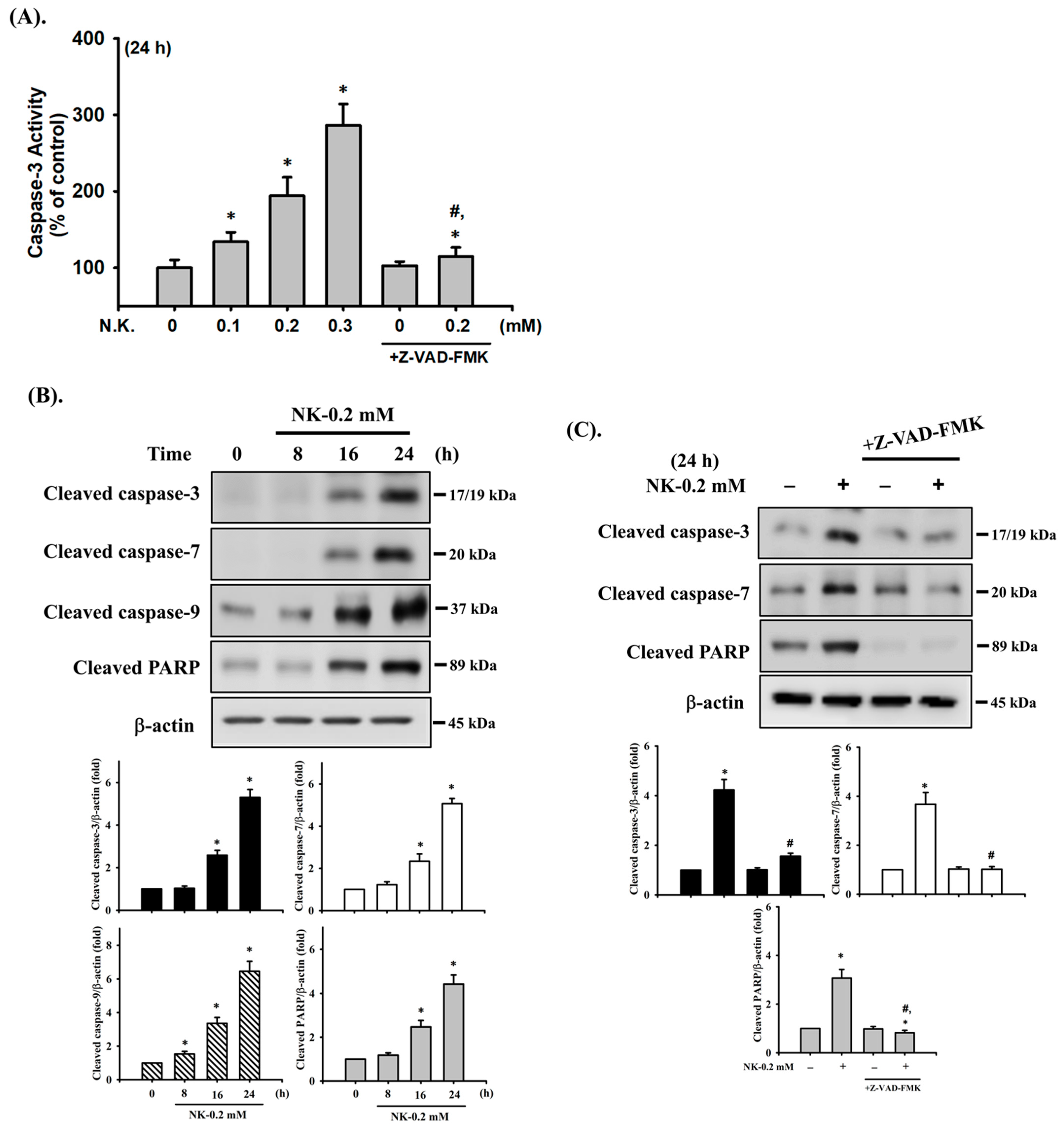
2.1. Effects of Ketamine and Its Metabolite Norketamine (NK) on Cell Viability and Apoptosis in RT4 Cells
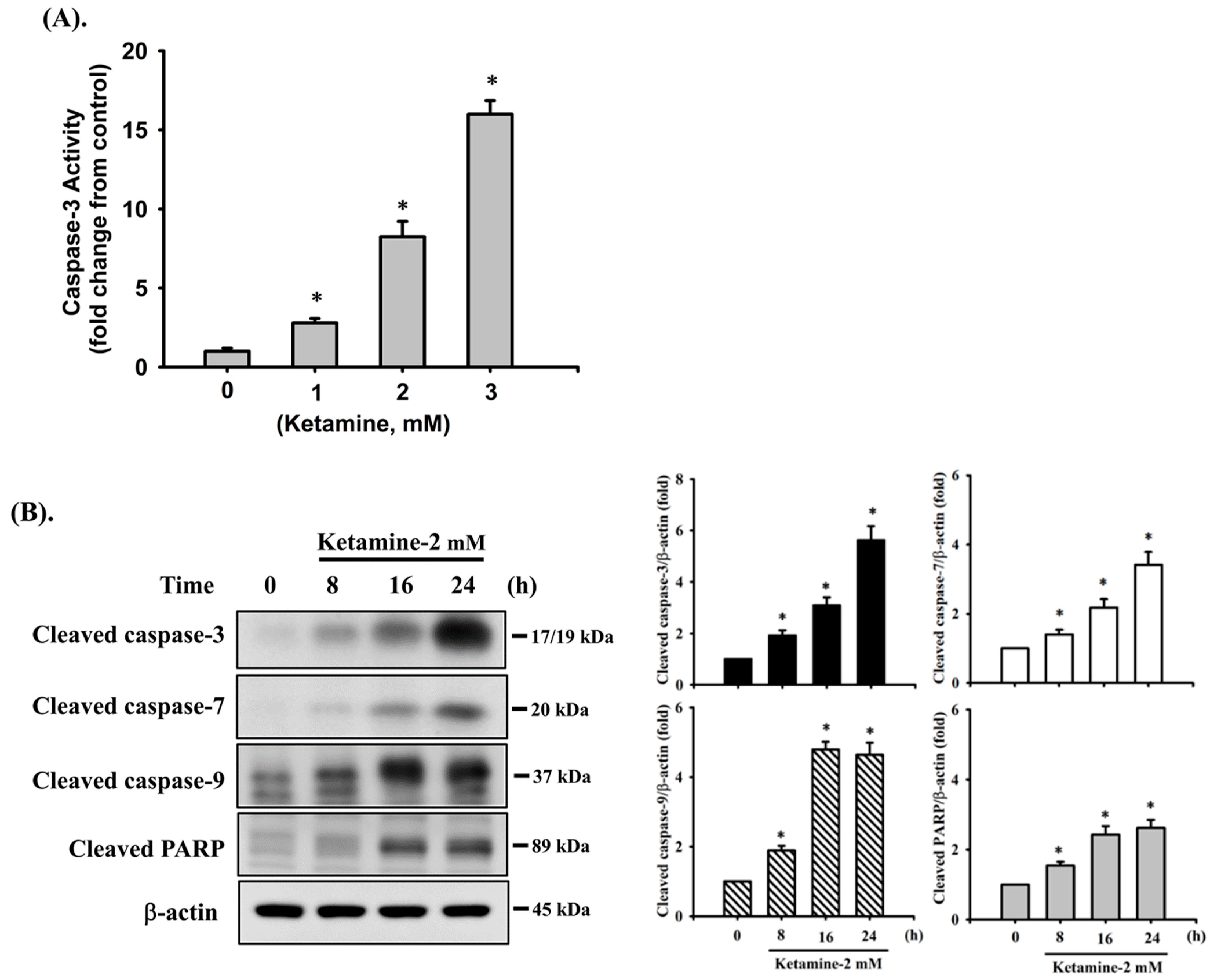
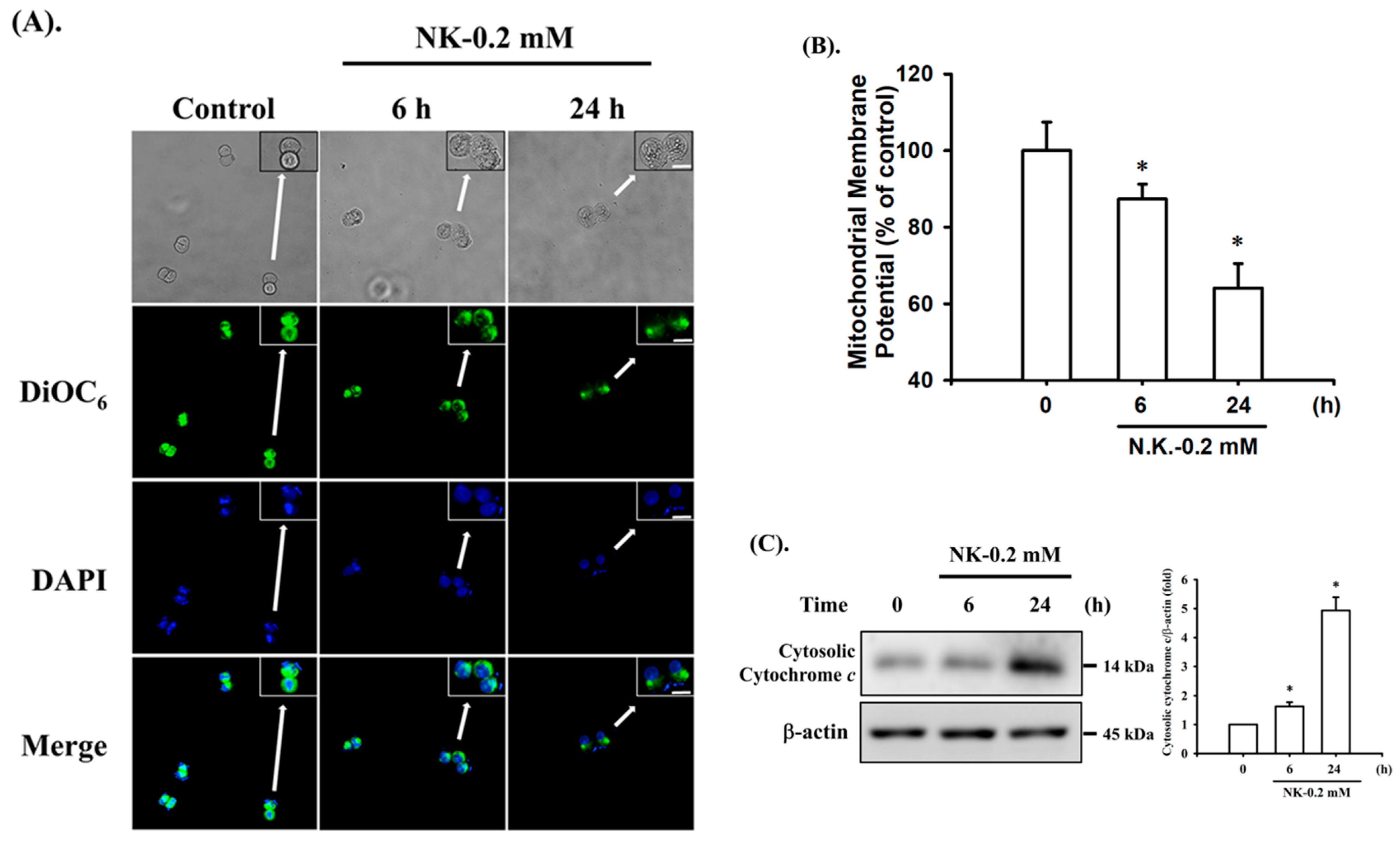
2.2. Norketamine (NK)-Induced Apoptosis Is Mediated by a Mitochondria-Dependent Pathway in RT4 Cells
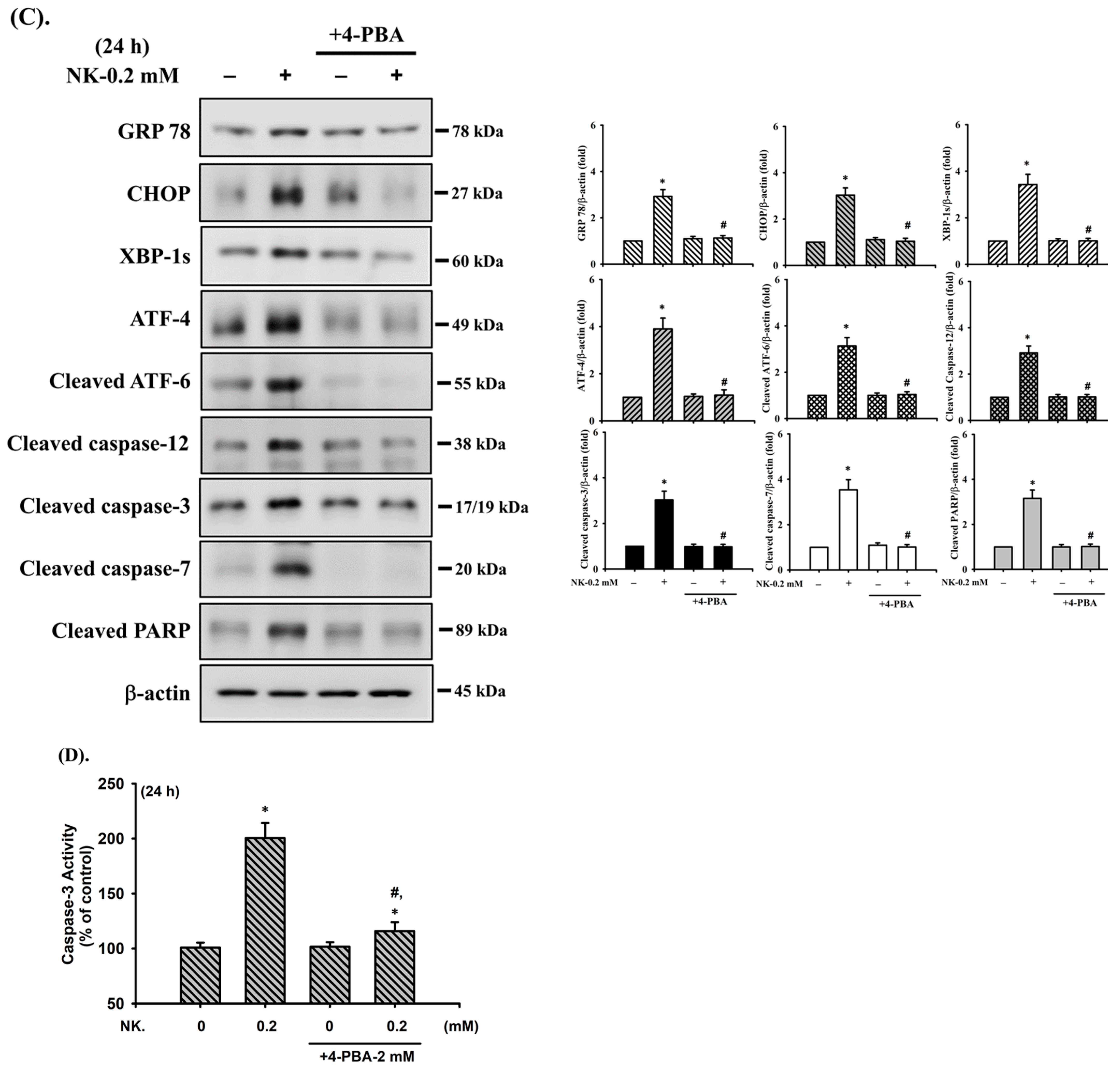
2.3. Norketamine (NK) Induces the ER Stress Response in RT4 Cells
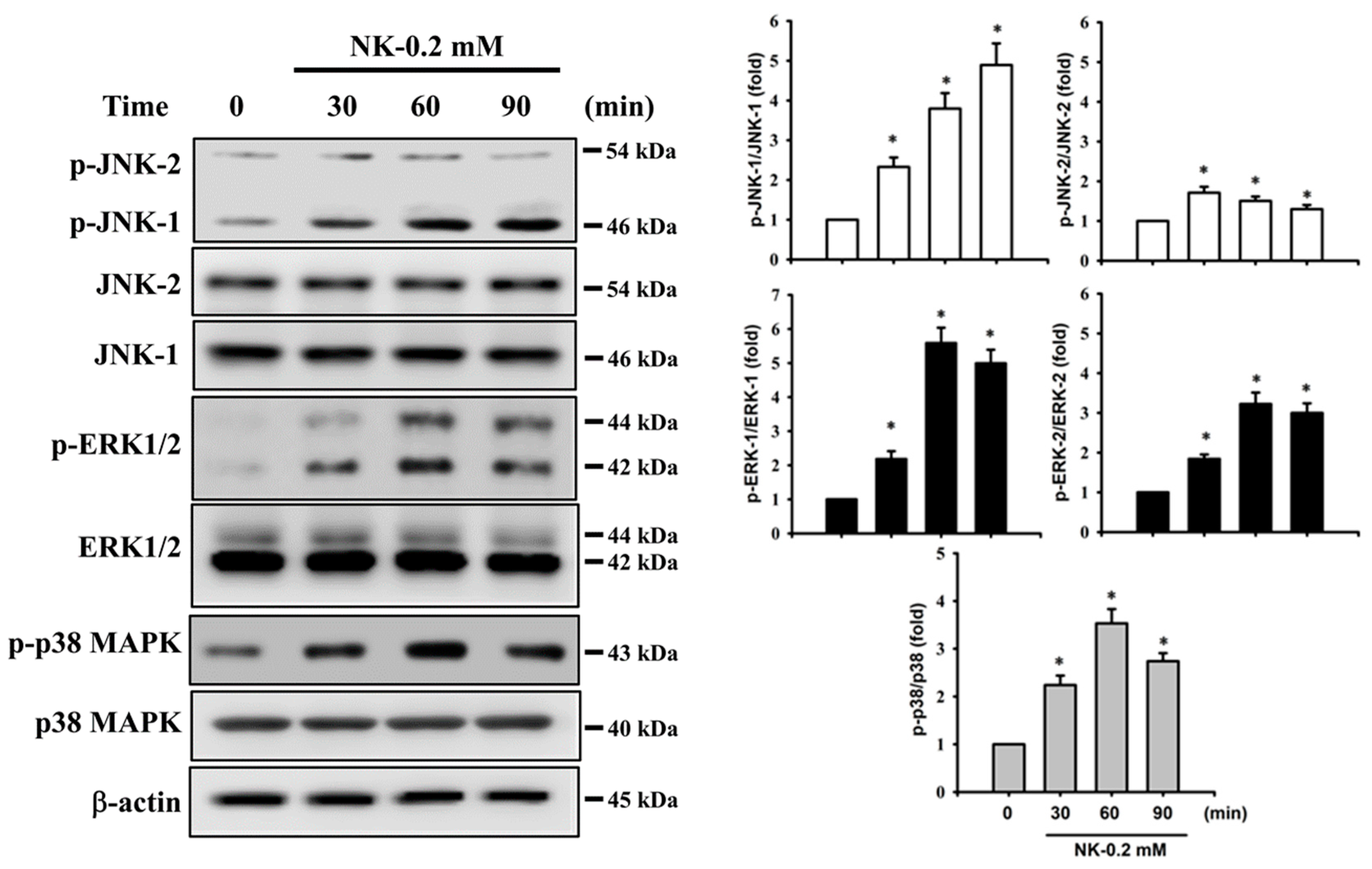
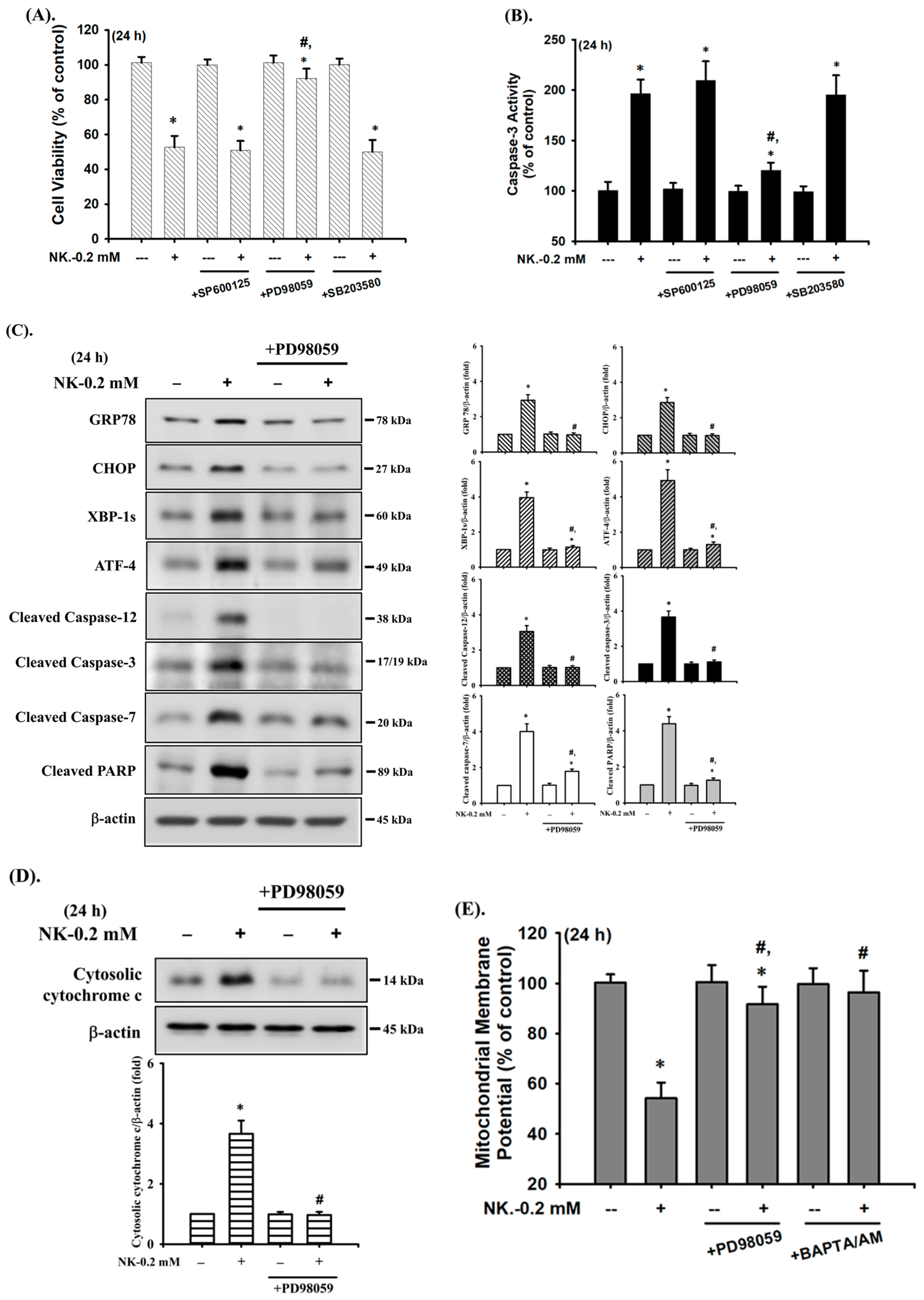
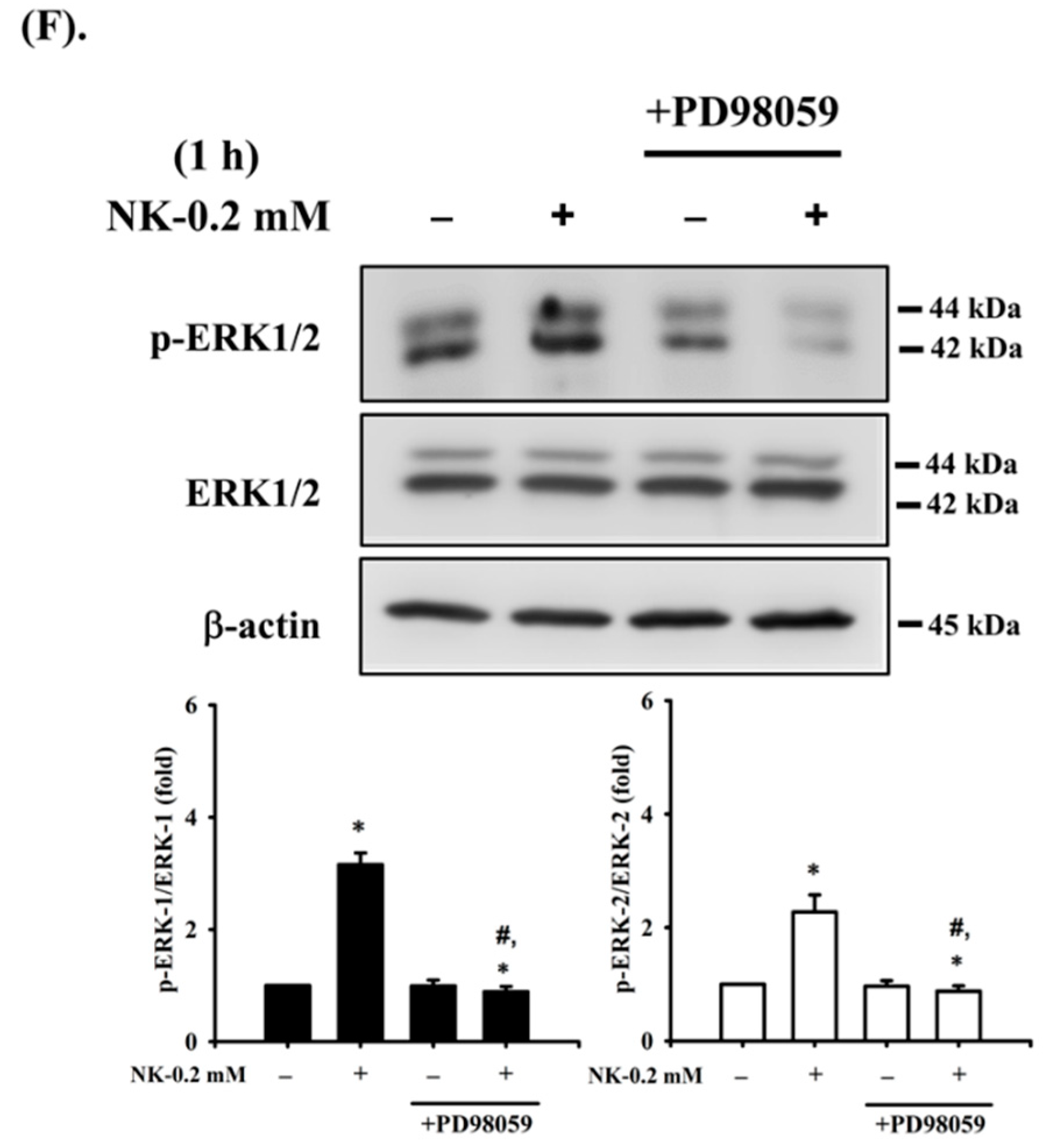
2.4. ERK1/2 Signaling Pathway Plays an Important Role in Norketamine (NK)-Induced RT4 Cell Apoptosis
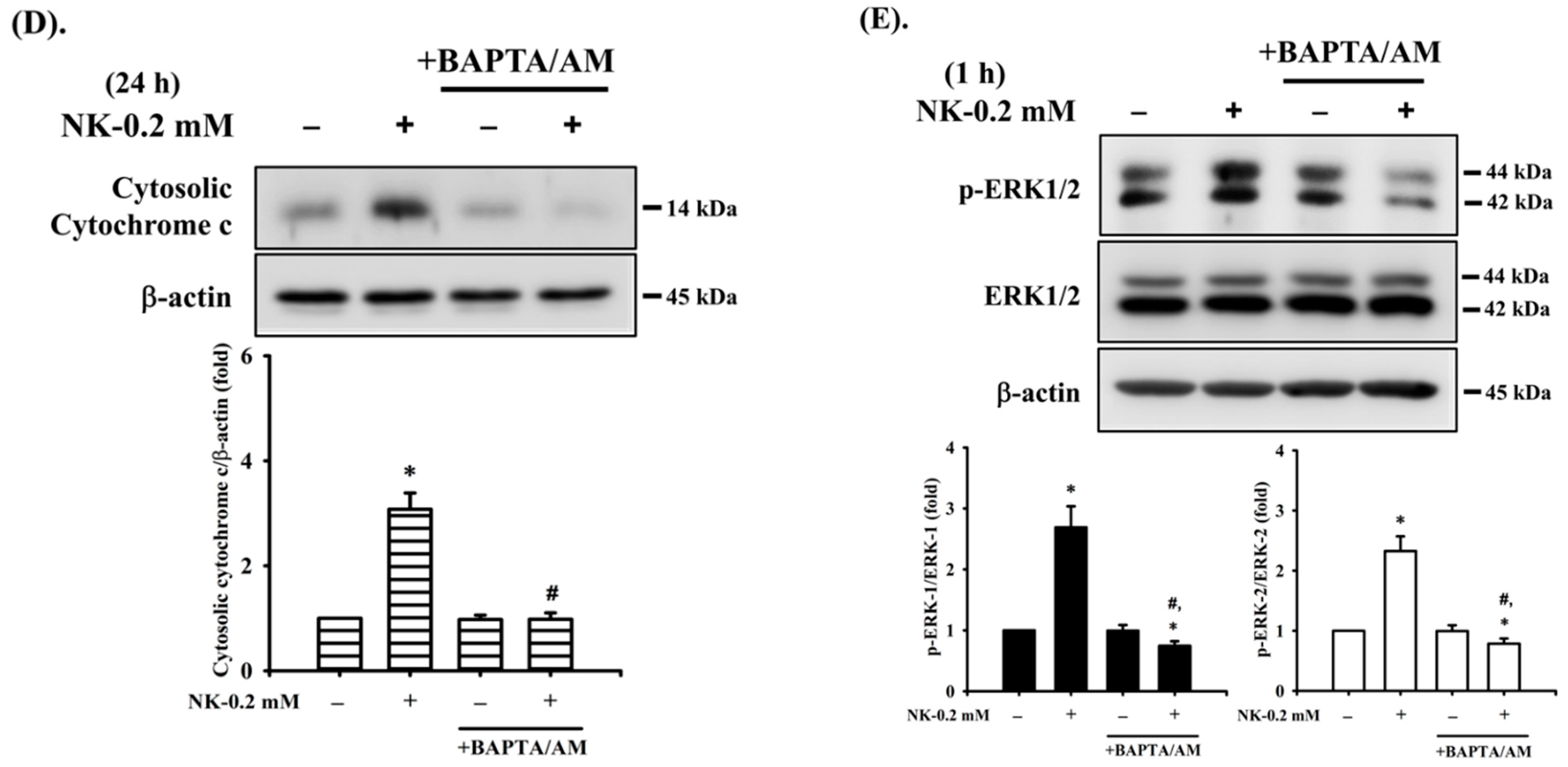
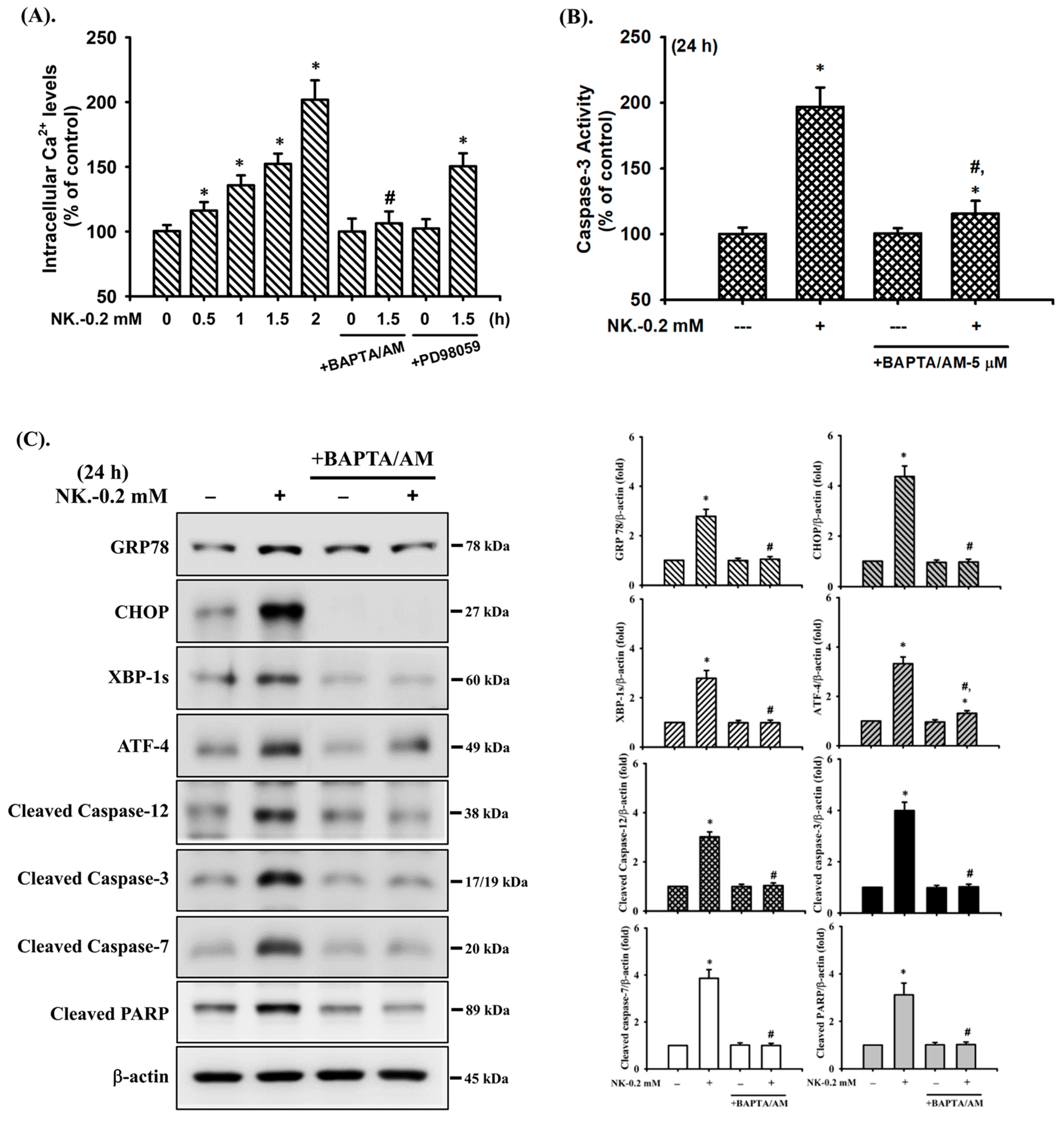
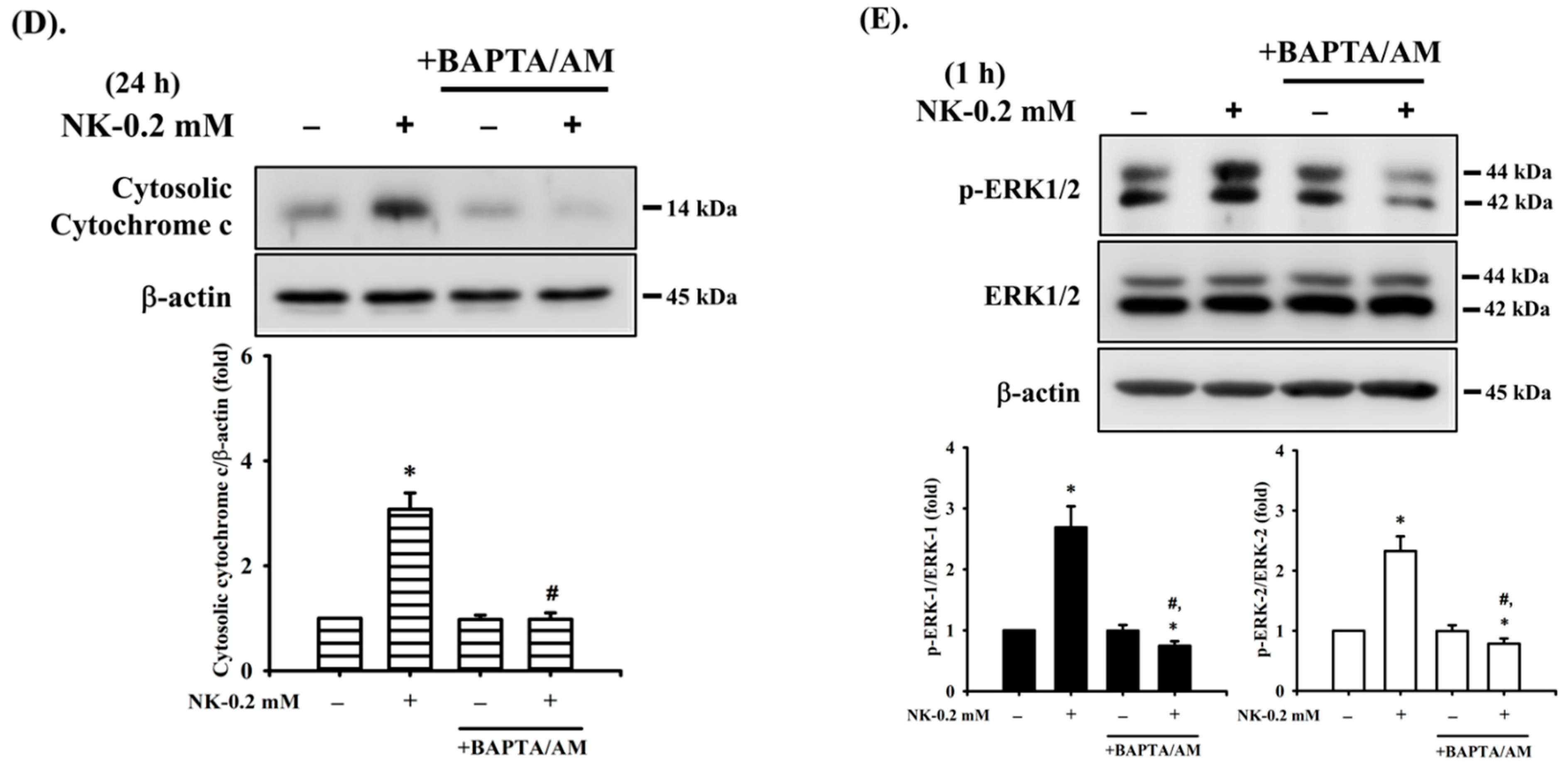
2.5. The Role of [Ca2+]i Signaling in Norketamine (NK)-Induced RT4 Cell Apoptosis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Determination of Caspase-3 Activity
4.5. Detection of Mitochondrial Membrane Potential (MMP)
4.6. Western Blot Analysis
4.7. Measurement of Intracellular Calcium Levels
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF | activating transcription factor |
| [Ca2+]i | calcium ion |
| CHOP | the CCAAT/enhancer-binding protein (C/EBP) homologous protein |
| eIF2α | eukaryotic translation initiation factor 2 subunit α |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| GRP | glucose regulated protein |
| IRE-1 | inositol-requiring enzyme 1 |
| MMP | mitochondrial membrane potential |
| NK | norketamine |
| 4-PBA | 4-phenylbutyric acid |
| PARP | poly(ADP-ribose) polymerase |
| PERK | protein kinase RNA-like endoplasmic reticulum kinase |
| UPR | unfolded protein response |
| XBP-1 | X-box binding protein-1 |
References
- Morgan, C.J.A.; Curran, H.V.; Independent Scientific Committee on Drugs (ISCD). Ketamine use: A review. Addiction 2012, 107, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wu, S.; Ni, L.; Chen, Z.; Li, X.; Yang, S.; Gui, Y.; Guan, Z.; Cai, Z.; Ye, J. Ketamine-Associated Urinary Tract Dysfunction: An Underrecognized Clinical Entity. Urol. Int. 2012, 89, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Lui, L.M.W.; Rosenblat, J.D.; Teopiz, K.M.; Lipsitz, O.; Cha, D.S.; Xiong, J.; Nasri, F.; Lee, Y.; Kratiuk, K.; et al. Ketamine-induced urological toxicity: Potential mechanisms and translation for adults with mood disorders receiving ketamine treatment. Psychopharmacology 2021, 238, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Shahani, R.; Streutker, C.; Dickson, B.; Stewart, R.J. Ketamine-Associated Ulcerative Cystitis: A New Clinical Entity. Urology 2007, 69, 810–812. [Google Scholar] [CrossRef]
- Baker, S.C.; Stahlschmidt, J.; Oxley, J.; Hinley, J.; Eardley, I.; Marsh, F.; Gillatt, D.; Fulford, S.; Southgate, J. Nerve hyperplasia: A unique feature of ketamine cystitis. Acta Neuropathol. Commun. 2013, 1, 64. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Wu, T.; Yi, X.; Liu, L.; Yan, J.; Lu, Z. Changes to the bladder epithelial barrier are associated with ketamine-induced cystitis. Exp. Ther. Med. 2017, 14, 2757–2762. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.C.; Shabir, S.; Georgopoulos, N.T.; Southgate, J. Ketamine-Induced Apoptosis in Normal Human Urothelial Cells: A Direct, N-Methyl-d-Aspartate Receptor–Independent Pathway Characterized by Mitochondrial Stress. Am. J. Pathol. 2016, 186, 1267–1277. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.; Wei, L.; Yu, S.; Jiang, S.; Ma, Y.; Zhang, C.; Wang, J.; Gao, Z.; Wan, F.; Zhuang, G.; et al. Ketamine induces reactive oxygen species and enhances autophagy in SV-HUC-1 human uroepithelial cells. J. Cell Physiol. 2019, 234, 2778–2787. [Google Scholar] [CrossRef]
- Liu, K.M.; Chuang, S.M.; Long, C.Y.; Lee, Y.L.; Wang, C.C.; Lu, M.C.; Lin, R.J.; Lu, J.H.; Jang, M.Y.; Wu, W.J.; et al. Ketamine-induced ulcerative cystitis and bladder apoptosis involve oxidative stress mediated by mitochondria and the endoplasmic reticulum. Am. J. Physiol. Ren. Physiol. 2015, 309, F318–F331. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wu, D.; Li, Y.; Qiao, H.; Shan, Z. Ketamine enhances autophagy and endoplasmic reticulum stress in rats and SV-HUC-1 cells via activating IRE1-TRAF2-ASK1-JNK pathway. Cell Cycle 2021, 20, 1907–1922. [Google Scholar] [CrossRef]
- Chan, W.H.; Sun, W.Z.; Ueng, T.H. Induction of rat hepatic cytochrome P-450 by ketamine and its toxicological implications. J. Toxicol. Environ. Health Part A 2005, 68, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Adamowicz, P.; Kala, M. Urinary excretion rates of ketamine and norketamine following therapeutic ketamine administration: Method and detection window considerations. J. Anal. Toxicol. 2005, 29, 376–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.A.; Sklerov, J.; Levine, B.; Jacobs, A.J. Urine concentrations of ketamine and norketamine following illegal consumption. J. Anal. Toxicol. 2001, 25, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.; Lin, C.C.; Lin, A.T.L.; Fan, Y.H.; Chen, K.K. Ketamine-Induced Uropathy: A New Clinical Entity Causing Lower Urinary Tract Symptoms. LUTS Low. Urin. Tract Symptoms 2012, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.L.; Jiang, Y.H.; Kuo, H.C. Increased apoptosis and suburothelial inflammation in patients with ketamine-related cystitis: A comparison with non-ulcerative interstitial cystitis and controls. BJU Int. 2013, 112, 1156–1162. [Google Scholar] [CrossRef]
- Krieger, C.; Duchen, M.R. Mitochondria, Ca2+ and neurodegenerative disease. Eur. J. Pharmacol. 2002, 447, 177–188. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5, 175. [Google Scholar] [CrossRef]
- Wu, P.; Shan, Z.; Wang, Q.; Huang, J.; Zheng, S.; Shan, Z. Involvement of Mitochondrial Pathway of Apoptosis in Urothelium in Ketamine-Associated Urinary Dysfunction. Am. J. Med. Sci. 2015, 349, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Healy, S.J.; Jager, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Yang, C.Y.; Su, C.C.; Fang, K.M.; Yen, C.C.; Lin, C.T.; Liu, J.M.; Lee, K.I.; Chen, Y.W.; Liu, S.H.; et al. 4-Methyl-2,4-bis(4-hydroxyphenyl)pent-1-ene, a Major Active Metabolite of Bisphenol A, Triggers Pancreatic β-Cell Death via a JNK/AMPKα Activation-Regulated Endoplasmic Reticulum Stress-Mediated Apoptotic Pathway. Int. J. Mol. Sci. 2021, 22, 4379. [Google Scholar] [CrossRef]
- Chen, S.; Melchior, W.B.; Guo, L. Endoplasmic Reticulum Stress in Drug- and Environmental Toxicant-Induced Liver Toxicity. J. Environ. Sci. Health Part C 2014, 32, 83–104. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.P.; Yen, C.C.; Tang, F.C.; Lee, K.I.; Liu, S.H.; Wu, C.C.; Hsieh, S.S.; Su, C.C.; Kuo, C.Y.; Chen, Y.W. Methylmercury exposure induces ROS/Akt inactivation-triggered endoplasmic reticulum stress-regulated neuronal cell apoptosis. Toxicology 2019, 425, 152245. [Google Scholar] [CrossRef]
- Karna, K.K.; Shin, Y.S.; Choi, B.R.; Kim, H.K.; Park, J.K. The Role of Endoplasmic Reticulum Stress Response in Male Reproductive Physiology and Pathology: A Review. World J. Men’s Health 2020, 38, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, S.; Agartz, I.; Ögren, S.O.; Patrone, C.; Lundberg, M. PACAP Protects Adult Neural Stem Cells from the Neurotoxic Effect of Ketamine Associated with Decreased Apoptosis, ER Stress and mTOR Pathway Activation. PLoS ONE 2017, 12, e0170496. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jiang, X.; Zhang, C.; Li, D.; Yu, S.; Wan, F.; Ma, Y.; Guo, W.; Shan, Z. Ketamine induces endoplasmic reticulum stress in rats and SV-HUC-1 human uroepithelial cells by activating NLRP3/TXNIP aix. Biosci. Rep. 2019, 39, BSR20190595. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, J.; Lim, W.; You, S.; Song, G. Butylated hydroxyanisole induces testicular dysfunction in mouse testis cells by dysregulating calcium homeostasis and stimulating endoplasmic reticulum stress. Sci. Total Environ. 2020, 702, 134775. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Kuo, C.Y.; Yang, C.Y.; Liu, J.M.; Hsu, R.J.; Lee, K.I.; Su, C.C.; Wu, C.C.; Lin, C.T.; Liu, S.H.; et al. Cadmium exposure induces pancreatic β-cell death via a Ca2+-triggered JNK/CHOP-related apoptotic signaling pathway. Toxicology 2019, 425, 152252. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.C.; Jivan, A.; Shao, C.; Duan, L.; Goad, D.; Zaganjor, E.; Osborne, J.; McGlynn, K.; Stippec, S.; Earnest, S.; et al. The roles of MAPKs in disease. Cell Res. 2008, 18, 436–442. [Google Scholar] [CrossRef]
- Liu, L.; Chang, X.; Zhang, Y.; Wu, C.; Li, R.; Tang, L.; Zhou, Z. Fluorochloridone induces primary cultured Sertoli cells apoptosis: Involvement of ROS and intracellular calcium ions-mediated ERK1/2 activation. Toxicol. In Vitro 2018, 47, 228–237. [Google Scholar] [CrossRef]
- Liu, W.; Xu, C.; Ran, D.; Wang, Y.; Zhao, H.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; Liu, Z. CaMKⅡ mediates cadmium induced apoptosis in rat primary osteoblasts through MAPK activation and endoplasmic reticulum stress. Toxicology 2018, 406-407, 70–80. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahar, E.; Kim, H.; Yoon, H.E.R. Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Loncke, J.; Kaasik, A.; Bezprozvanny, I.; Parys, J.B.; Kerkhofs, M.; Bultynck, G. Balancing ER-Mitochondrial Ca2+ Fluxes in Health and Disease. Trends Cell Biol. 2021, 31, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.T.; Huang, K.H.; Chiang, J.Y.; Sung, P.H.; Huang, C.R.; Chu, Y.C.; Chuang, F.C.; Yip, H.K. Extracorporeal Shock Wave Therapy Protected the Functional and Architectural Integrity of Rodent Urinary Bladder against Ketamine-Induced Damage. Biomedicines 2021, 9, 1391. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.H.; Wu, Y.H.; Juan, T.J.; Lin, H.Y.; Lin, R.J.; Chueh, K.S.; Lee, Y.C.; Chang, C.Y.; Juan, Y.S. Autophagy Alters Bladder Angiogenesis and Improves Bladder Hyperactivity in the Pathogenesis of Ketamine-Induced Cystitis in a Rat Model. Biology 2021, 10, 488. [Google Scholar] [CrossRef] [PubMed]
- Zuo, D.; Sun, F.; Cui, J.; Liu, Y.; Liu, Z.; Zhou, X.; Li, Z.; Wu, Y. Alcohol amplifies ketamine-induced apoptosis in primary cultured cortical neurons and PC12 cells through down-regulating CREB-related signaling pathways. Sci. Rep. 2017, 7, 10523. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xu, G.; Huang, M.; Fu, L.; Jiang, X.; Yang, M. Bisphenol A and bisphenol AF co-exposure induces apoptosis in human granulosa cell line KGN through intracellular stress-dependent mechanisms. Arab. J. Chem. 2021, 14, 103399. [Google Scholar] [CrossRef]
- Domino, E.F. Taming the ketamine tiger. 1965. Anesthesiology 2010, 113, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Lankenau, S.E.; Clatts, M.C. Drug injection practices among high-risk youths: The first shot of ketamine. J. Urban Health 2004, 81, 232–248. [Google Scholar] [CrossRef]
- Jhang, J.F.; Hsu, Y.H.; Kuo, H.C. Possible pathophysiology of ketamine-related cystitis and associated treatment strategies. Int. J. Urol. 2015, 22, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Chen, R.M. Mechanisms of ketamine-involved regulation of cytochrome P450 gene expression. Expert Opin. Drug Metab. Toxicol. 2010, 6, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Juan, Y.S.; Lee, Y.L.; Long, C.Y.; Wong, J.H.; Jang, M.Y.; Lu, J.H.; Wu, W.J.; Huang, Y.S.; Chang, W.C.; Chuang, S.M. Translocation of NF-κB and Expression of Cyclooxygenase-2 Are Enhanced by Ketamine-Induced Ulcerative Cystitis in Rat Bladder. Am. J. Pathol. 2015, 185, 2269–2285. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, M.; Leeuwenburgh, C. Apoptosis and Aging: Role of the Mitochondria. J. Gerontol. Ser. A 2001, 56, B475–B482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.L.; Chen, T.; Wong, Y.S.; Xu, G.; Fan, R.R.; Zhao, H.L.; Chan, J.C.N. Involvement of mitochondrial dysfunction in human islet amyloid polypeptide-induced apoptosis in INS-1E pancreatic beta cells: An effect attenuated by phycocyanin. Int. J. Biochem. Cell Biol. 2011, 43, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Vela-Guajardo, J.E.; Garza-González, S.; García, N. Glucolipotoxicity-induced Oxidative Stress is Related to Mitochondrial Dysfunction and Apoptosis of Pancreatic β-cell. Curr. Diabetes Rev. 2021, 17, 46–56. [Google Scholar] [CrossRef]
- Yang, T.Y.; Yen, C.C.; Lee, K.I.; Su, C.C.; Yang, C.Y.; Wu, C.C.; Hsieh, S.S.; Ueng, K.C.; Huang, C.F. Molybdenum induces pancreatic β-cell dysfunction and apoptosis via interdependent of JNK and AMPK activation-regulated mitochondria-dependent and ER stress-triggered pathways. Toxicol. Appl. Pharmacol. 2016, 294, 54–64. [Google Scholar] [CrossRef]
- Fu, S.C.; Liu, J.M.; Lee, K.I.; Tang, F.C.; Fang, K.M.; Yang, C.Y.; Su, C.C.; Chen, H.H.; Hsu, R.J.; Chen, Y.W. Cr(VI) induces ROS-mediated mitochondrial-dependent apoptosis in neuronal cells via the activation of Akt/ERK/AMPK signaling pathway. Toxicol. In Vitro 2020, 65, 104795. [Google Scholar] [CrossRef]
- Lu, T.H.; Tseng, T.J.; Su, C.C.; Tang, F.C.; Yen, C.C.; Liu, Y.Y.; Yang, C.Y.; Wu, C.C.; Chen, K.L.; Hung, D.Z.; et al. Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicol. Lett. 2014, 224, 130–140. [Google Scholar] [CrossRef]
- Bai, X.; Yan, Y.; Canfield, S.; Muravyeva, M.Y.; Kikuchi, C.; Zaja, I.; Corbett, J.A.; Bosnjak, Z.J. Ketamine enhances human neural stem cell proliferation and induces neuronal apoptosis via reactive oxygen species-mediated mitochondrial pathway. Anesth. Analg. 2013, 116, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.T.; Wu, T.T.; Yu, P.Y.; Chen, R.M. Apoptotic insults to human HepG2 cells induced by S-(+)-ketamine occurs through activation of a Bax-mitochondria-caspase protease pathway. Br. J. Anaesth. 2009, 102, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.F.; Liu, S.H.; Su, C.C.; Fang, K.M.; Yen, C.C.; Yang, C.Y.; Tang, F.C.; Liu, J.M.; Wu, C.C.; Lee, K.I.; et al. Roles of ERK/Akt signals in mitochondria-dependent and endoplasmic reticulum stress-triggered neuronal cell apoptosis induced by 4-methyl-2,4-bis(4-hydroxyphenyl)pent-1-ene, a major active metabolite of bisphenol A. Toxicology 2021, 455, 152764. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009, 16, 368–377. [Google Scholar] [CrossRef]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res. 2018, 138, 99–142. [Google Scholar] [CrossRef]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017, 284, 4177–4195. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Du, L.; Cao, J.; Hölscher, C.; Feng, Y.; Su, H.; Wang, Y.; Yun, K.M. Levo-tetrahydropalmatine inhibits the acquisition of ketamine-induced conditioned place preference by regulating the expression of ERK and CREB phosphorylation in rats. Behav. Brain Res. 2017, 317, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Li, X.; Jia, S.; Liu, S.; Fu, L.; Jiang, X.; Yang, M. Bisphenol AF induces apoptosis via estrogen receptor beta (ERβ) and ROS-ASK1-JNK MAPK pathway in human granulosa cell line KGN. Environ. Pollut. 2021, 270, 116051. [Google Scholar] [CrossRef]
- Liu, W.; Yang, T.; Xu, Z.; Xu, B.; Deng, Y. Methyl-mercury induces apoptosis through ROS-mediated endoplasmic reticulum stress and mitochondrial apoptosis pathways activation in rat cortical neurons. Free Radic. Res. 2019, 53, 26–44. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.; Acosta, L.; Karadayian, A.G.; Lores-Arnaiz, S. Ketamine induced cell death can be mediated by voltage dependent calcium channels in PC12 cells. Exp. Mol. Pathol. 2019, 111, 104318. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Matsuo, T.; Mitsunari, K.; Asai, A.; Ohba, K.; Sakai, H. A Review of Oxidative Stress and Urinary Dysfunction Caused by Bladder Outlet Obstruction and Treatments Using Antioxidants. Antioxidants 2019, 8, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, X.J.; Zeng, J.J.; Lu, Y.; Chen, S.H.; Jiang, Z.W.; He, P.J.; Mi, H. Extracellular vesicles enhance oxidative stress through P38/NF-kB pathway in ketamine-induced ulcerative cystitis. J. Cell. Mol. Med. 2020, 24, 7609–7624. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Habeos, I.G.; Samuelson, A.V.; Bohmann, D. The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. Publ. Coop. Univ. Tex. MD Anderson Cancer Cent. 2009, 48, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, D.N.; Jena, G.B. Effect of melatonin on the expression of Nrf2 and NF-κB during cyclophosphamide-induced urinary bladder injury in rat. J. Pineal Res. 2010, 48, 324–331. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Chen, W.; Eblin, K.E.; Gandolfi, J.A.; Zhang, D.D. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol. Appl. Pharmacol. 2007, 225, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Zhao, Z.; Lang, J.; Sun, B.; Zhao, Q. Nrf2 loss of function exacerbates endoplasmic reticulum stress-induced apoptosis in TBI mice. Neurosci. Lett. 2022, 770, 136400. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.-W.; Lin, Y.-C.; Liu, J.-M.; Liu, S.-H.; Fang, K.-M.; Hsu, R.-J.; Huang, C.-F.; Chang, K.-Y.; Lee, K.-I.; Chang, K.-C.; et al. Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway. Int. J. Mol. Sci. 2022, 23, 4666. https://doi.org/10.3390/ijms23094666
Lin J-W, Lin Y-C, Liu J-M, Liu S-H, Fang K-M, Hsu R-J, Huang C-F, Chang K-Y, Lee K-I, Chang K-C, et al. Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway. International Journal of Molecular Sciences. 2022; 23(9):4666. https://doi.org/10.3390/ijms23094666
Chicago/Turabian StyleLin, Jhe-Wei, Yi-Chun Lin, Jui-Ming Liu, Shing-Hwa Liu, Kai-Min Fang, Ren-Jun Hsu, Chun-Fa Huang, Kai-Yao Chang, Kuan-I Lee, Kai-Chih Chang, and et al. 2022. "Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway" International Journal of Molecular Sciences 23, no. 9: 4666. https://doi.org/10.3390/ijms23094666
APA StyleLin, J.-W., Lin, Y.-C., Liu, J.-M., Liu, S.-H., Fang, K.-M., Hsu, R.-J., Huang, C.-F., Chang, K.-Y., Lee, K.-I., Chang, K.-C., Su, C.-C., & Chen, Y.-W. (2022). Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway. International Journal of Molecular Sciences, 23(9), 4666. https://doi.org/10.3390/ijms23094666