
Novel Multifaceted Roles for RNF213 Protein

 , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. RNF213 from Gene to Molecular Structure
- −
- Six ATPase domains, each containing a Walker A and a Walker B domain, peculiar to ATP-binding proteins; the A motif is the main ‘P-loop’ responsible for binding phosphate, while the B motif is a magnesium-binding motif [5].
- −
- One RING finger domain with an E3 ubiquitin ligase domain that is able to covalently modify the substrate with the addition of ubiquitin molecules and stimulate the intracellular biological processes, such as proteasomal protein degradation [4].
3. RNF213 Genetic Background
4. RNF213 and Inflammation-Related Angiogenesis
5. RNF213 Key Interactors
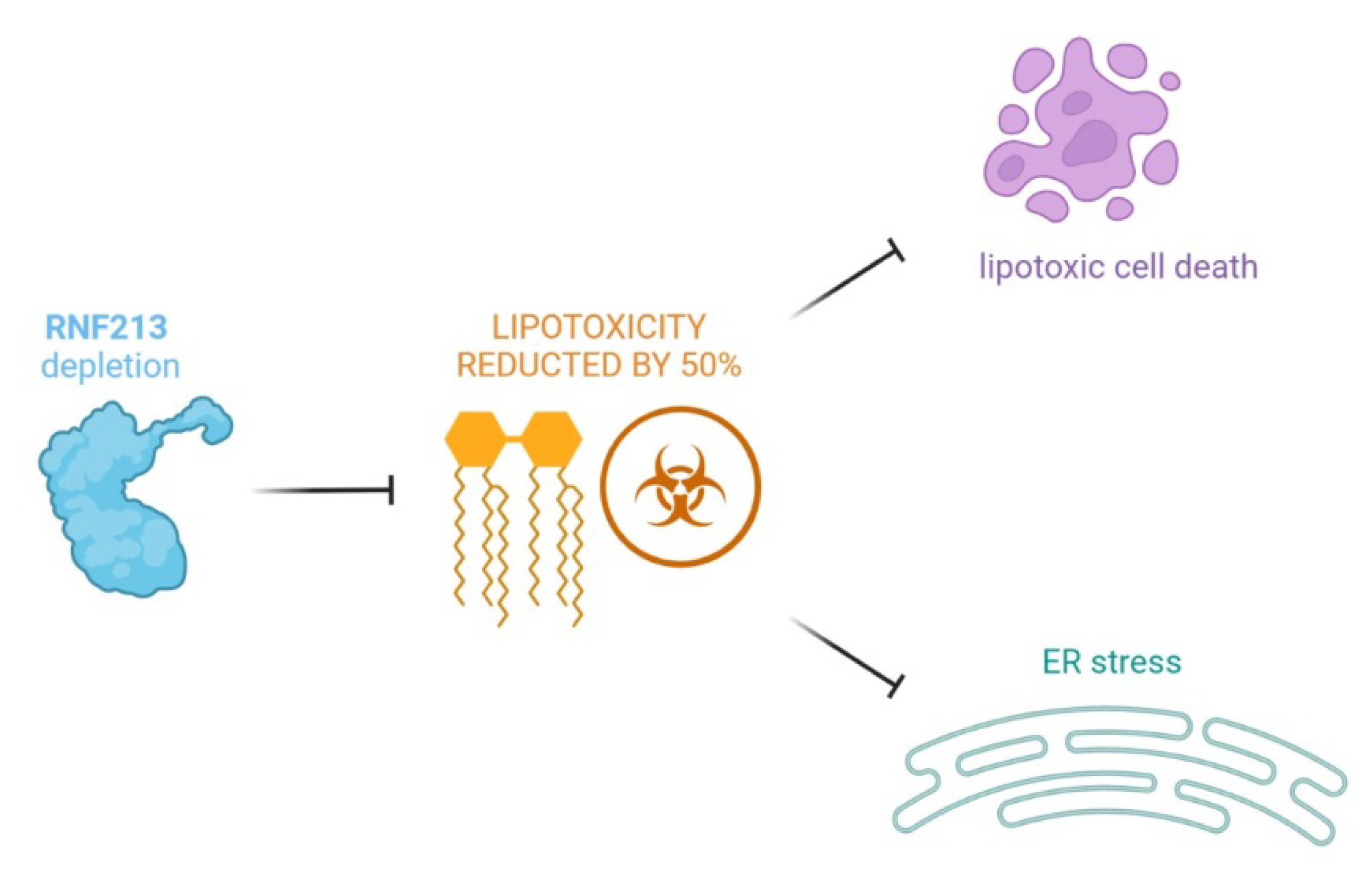
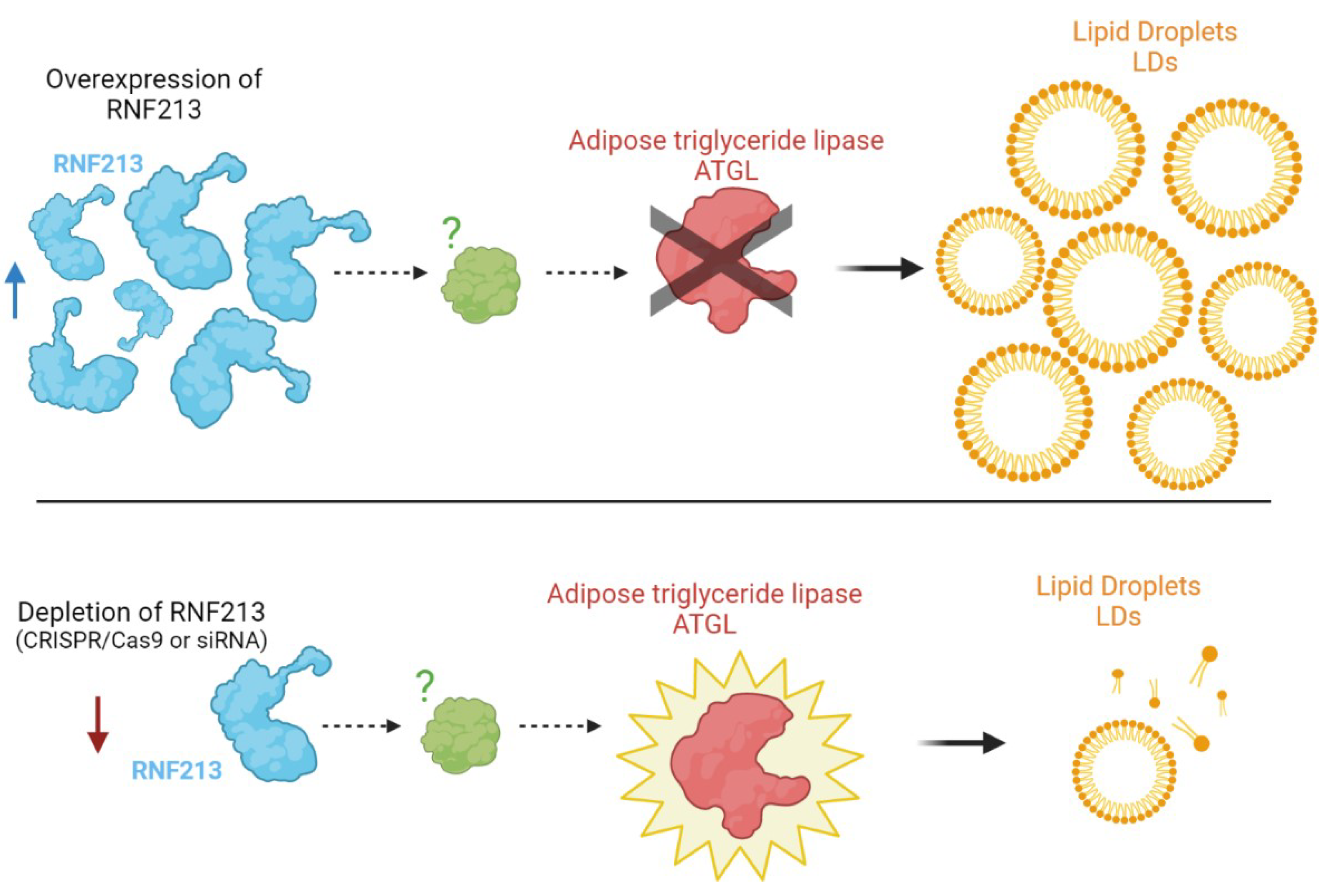
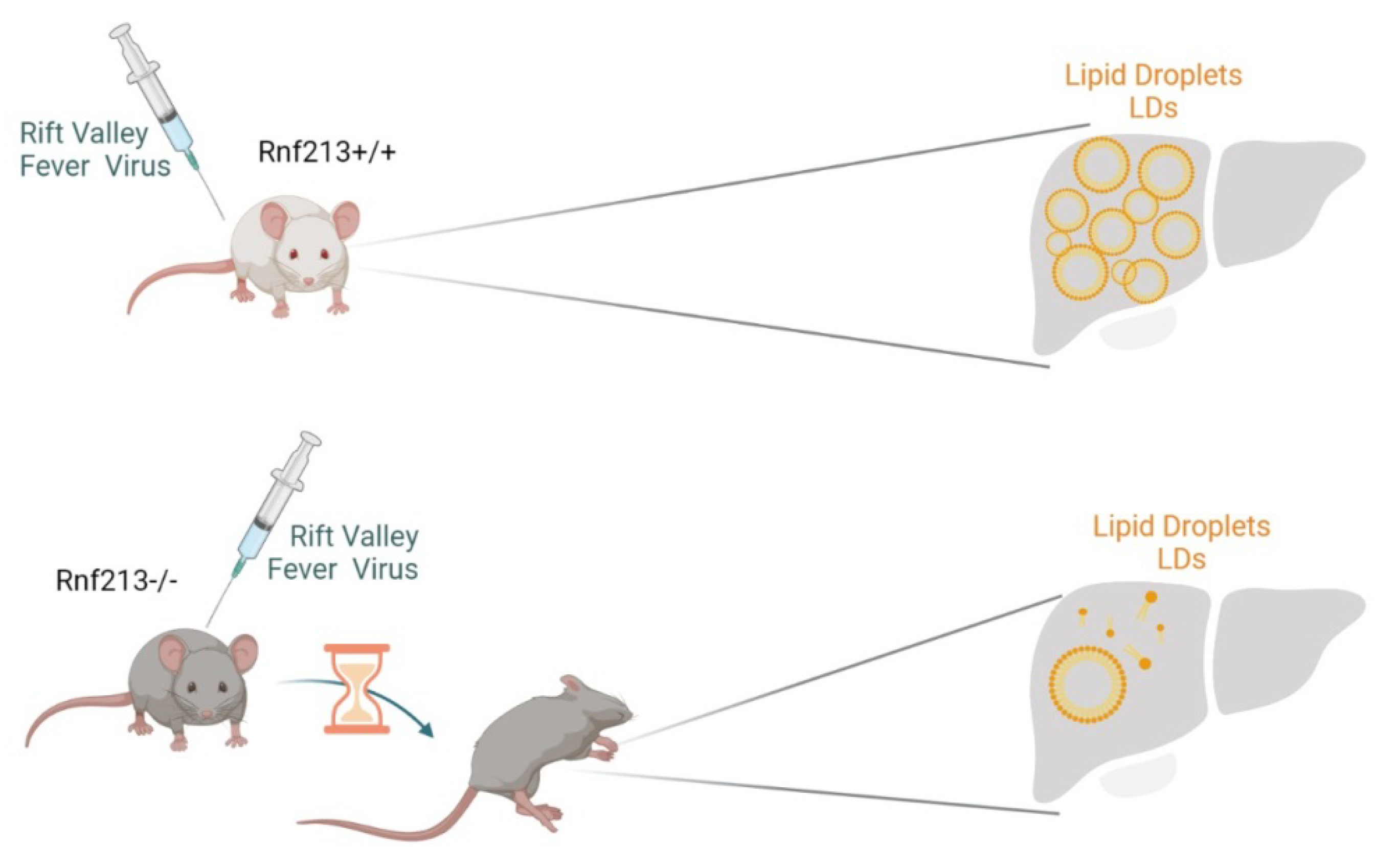
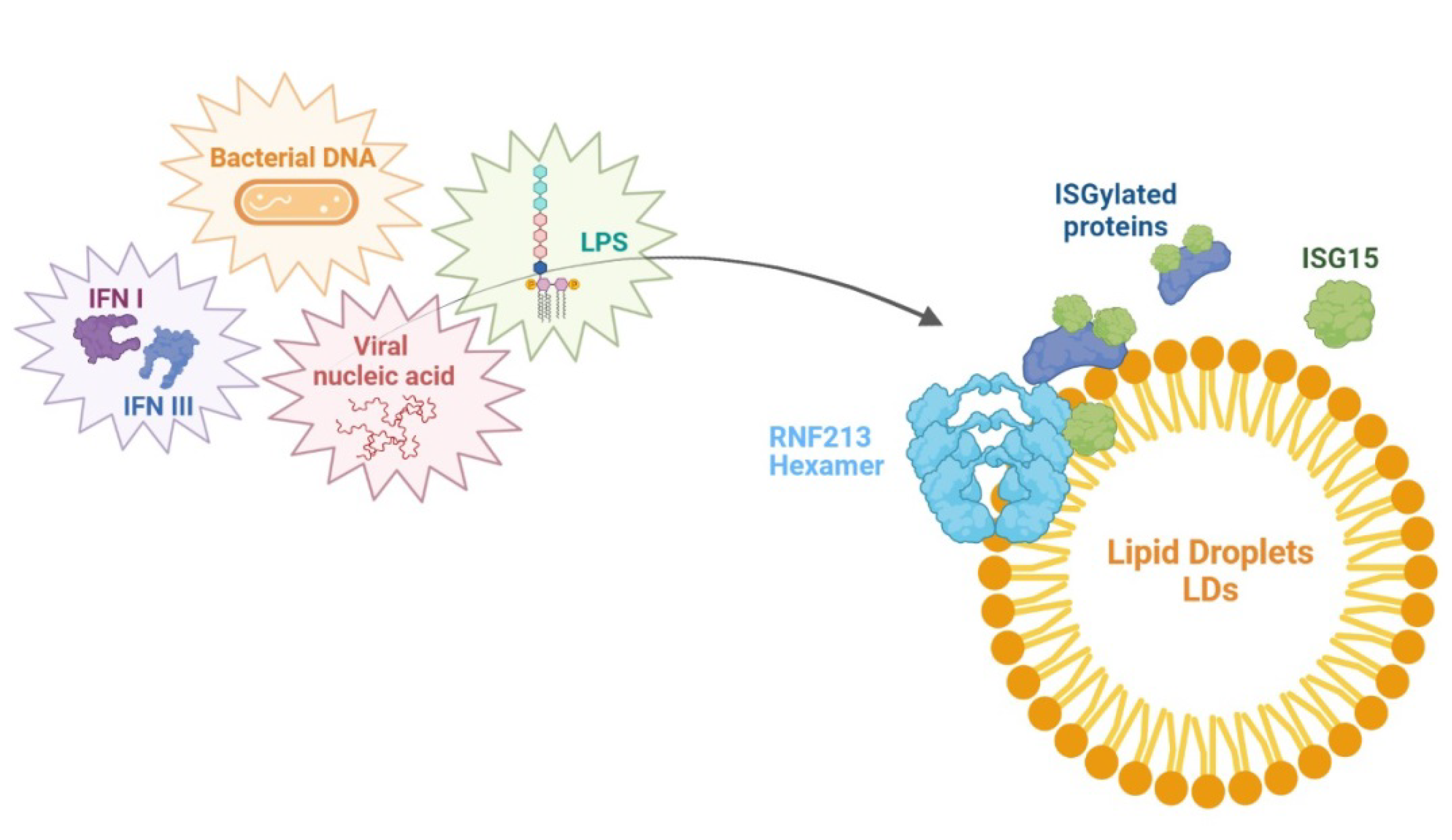
6. RNF213 and Lipid Metabolism
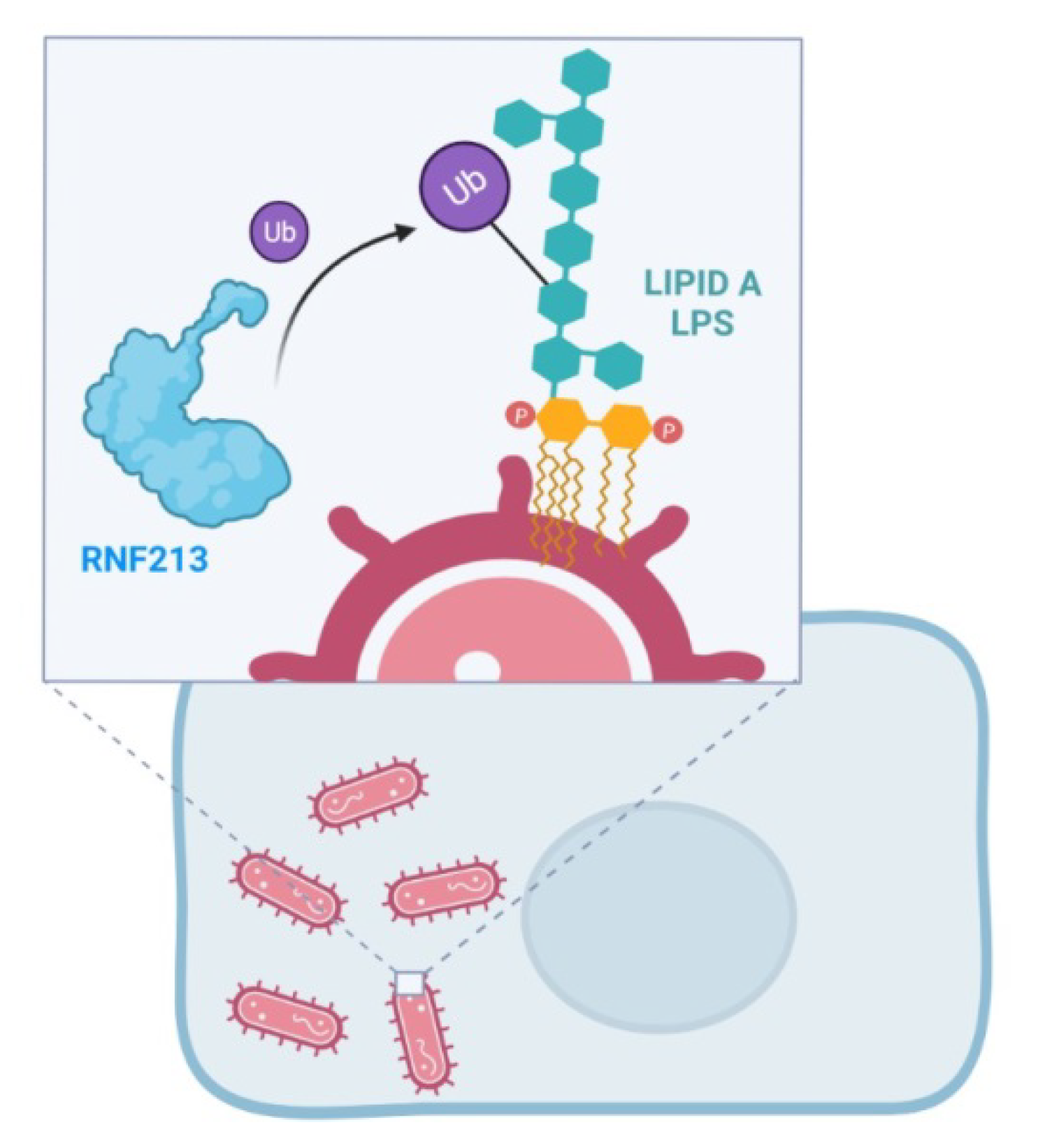
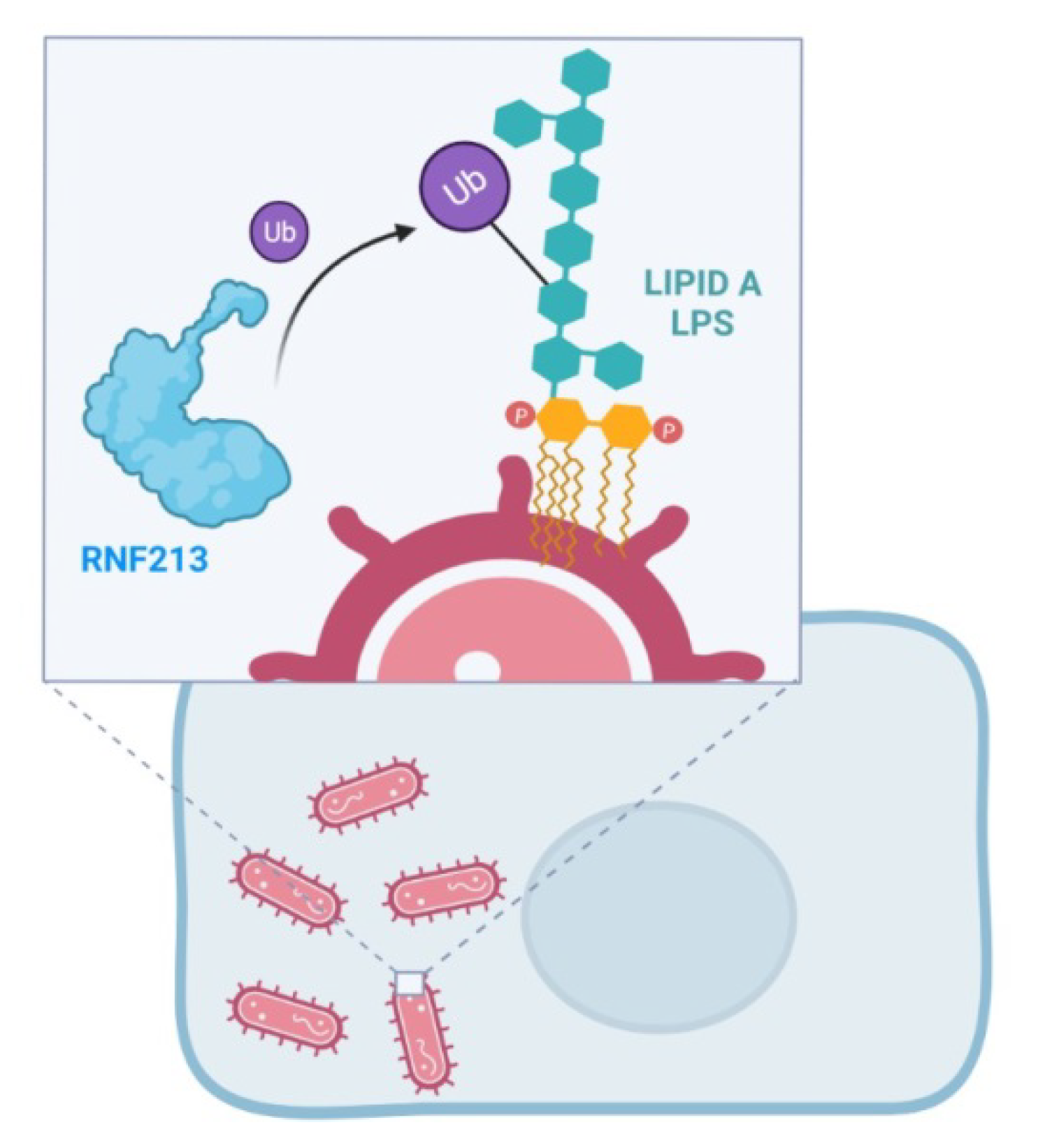
7. RNF213 and Antimicrobial Activity
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Ahel, J.; Lehner, A.; Vogel, A.; Schleiffer, A.; Meinhart, A.; Haselbach, D.; Clausen, T. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. eLife 2020, 9, 1–23. [Google Scholar] [CrossRef]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guey, S.; Kraemer, M.; Hervé, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, L.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef]
- Morito, D.; Nishikawa, K.; Hoseki, J.; Kitamura, A.; Kotani, Y.; Kiso, K.; Kinjo, M.; Fujiyoshi, Y.; Nagata, K. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci. Rep. 2014, 4, 4442. [Google Scholar] [CrossRef] [Green Version]
- Romero-Barrios, N.; Vert, G. Proteasome-independent functions of lysine-63 polyubiquitination in plants. New Phytol. 2018, 217, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, P.; Thirumurugan, K. New insights into TNFα/PTP1B and PPARγ pathway through RNF213- a link between inflammation, obesity, insulin resistance, and Moyamoya disease. Gene 2021, 771, 145340. [Google Scholar] [CrossRef]
- Liu, W.; Senevirathna, S.T.; Hitomi, T.; Kobayashi, H.; Roder, C.; Herzig, R.; Kraemer, M.; Voormolen, M.H.; Cahová, P.; Krischek, B.; et al. Genomewide association study identifies no major founder variant in Caucasian moyamoya disease. J. Genet. 2013, 92, 605–609. [Google Scholar] [CrossRef]
- Grangeon, L.; Guey, S.; Schwitalla, J.C.; Bergametti, F.; Arnould, M.; Corpechot, M.; Hadjadj, J.; Riant, F.; Aloui, C.; Drunat, S.; et al. Clinical and Molecular Features of 5 European Multigenerational Families with Moyamoya Angiopathy. Stroke 2019, 50, 789–796. [Google Scholar] [CrossRef]
- Guey, S.; Grangeon, L.; Brunelle, F.; Bergametti, F.; Amiel, J.; Lyonnet, S.; Delaforge, A.; Arnould, M.; Desnous, B.; Bellesme, C.; et al. De novo mutations in CBL causing early-onset paediatric moyamoya angiopathy. J. Med. Genet. 2017, 54, 550–557. [Google Scholar] [CrossRef]
- Kobayashi, H.; Matsuda, Y.; Hitomi, T.; Okuda, H.; Shioi, H.; Matsuda, T.; Imai, H.; Sone, M.; Taura, D.; Harada, K.H. Biochemical and Functional Characterization of RNF213 (Mysterin) R4810K, a Susceptibility Mutation of Moyamoya Disease, in Angiogenesis In Vitro and In Vivo. J. Am. Heart Assoc. 2015, 4, e002146. [Google Scholar] [CrossRef] [Green Version]
- Ohkubo, K.; Sakai, Y.; Inoue, H.; Akamine, S.; Ishizaki, Y.; Matsushita, Y.; Sanefuji, M.; Torisu, H.; Ihara, K.; Sardiello, M.; et al. Moyamoya disease susceptibility gene RNF213 links inflammatory and angiogenic signals in endothelial cells. Sci. Rep. 2015, 5, 13191. [Google Scholar] [CrossRef] [Green Version]
- Takagi, Y.; Kikuta, K.; Nozaki, K.; Fujimoto, M.; Hayashi, J.; Imamura, H.; Hashimoto, N. Expression of hypoxia-inducing factor-1 alpha and endoglin in intimal hyperplasia of the middle cerebral artery of patients with moyamoya disease. Neurosurgery 2007, 60, 338–345. [Google Scholar] [CrossRef]
- Takekawa, Y.; Umezawa, T.; Ueno, Y.; Sawada, T.; Kobayashi, M. Pathological and immunohistochemical findings of an autopsy case of adult moyamoya disease. Neuropathology 2004, 24, 236–242. [Google Scholar] [CrossRef]
- Takagi, Y.; Kikuta, K.; Nozaki, K.; Hashimoto, N. Histological features of middle cerebral arteries from patients treated for moyamoya disease. Neurol. Med. Chir. 2007, 47, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.J.; Bhattacharjee, M.B.; Regalado, E.S.; Milewicz, A.; El-Hakam, L.M.; Dauser, R.C.; Milewicz, D.M. Diffuse and uncontrolled vascular smooth muscle cell proliferation in rapidly progressing pediatric moyamoya disease. J. Neurosurg. Pediatr. 2010, 6, 244–249. [Google Scholar] [CrossRef]
- Achrol, A.S.; Guzman, R.; Lee, M.; Steinberg, G.K. Pathophysiology and genetic factors in moyamoya disease. Neurosurg. Focus. 2009, 26, E4. [Google Scholar] [CrossRef] [Green Version]
- Phi, J.H.; Suzuki, N.; Moon, Y.J.; Park, A.K.; Wang, K.C.; Lee, J.Y.; Choi, S.A.; Chong, S.; Shirane, R.; Kim, S.K. Chemokine Ligand 5 (CCL5) Derived from Endothelial Colony-Forming Cells (ECFCs) Mediates Recruitment of Smooth Muscle Progenitor Cells (SPCs) toward Critical Vascular Locations in Moyamoya Disease. PLoS ONE 2017, 12, e0169714. [Google Scholar] [CrossRef]
- Hamauchi, S.; Shichinohe, H.; Uchino, H.; Yamaguchi, S.; Nakayama, N.; Kazumata, K.; Osanai, T.; Abumiya, T.; Houkin, K.; Era, T. Cellular functions and gene and protein expression profiles in endothelial cells derived from Moyamoya disease-specific iPS cells. PLoS ONE 2016, 11, e163561. [Google Scholar] [CrossRef]
- Matsuo, M.; Nadanaka, S.; Soga, M.; Sugiyama, T.; Serigano, S.; Shimano, K.; Ichinose, F.; Nakamura, T.; Maeda, T.; Houkin, K. Vulnerability to shear stress caused by altered peri—Endothelial matrix is a key feature of Moyamoya disease. Sci. Rep. 2021, 11, 1552. [Google Scholar] [CrossRef] [PubMed]
- Habu, T.; Harada, K.H. UBC13 is an RNF213-associated E2 ubiquitin-conjugating enzyme, and Lysine 63-linked ubiquitination by the RNF213-UBC13 axis is responsible for angiogenic activity. FASEB Bio. Adv. 2021, 3, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gao, Y.; Li, L.; Jin, G.; Cai, Z.; Chao, J.I.; Lin, H.K. K63-linked ubiquitination in kinase activation and cancer. Front. Oncol. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickart, C.M.; Fushman, D. Polyubiquitin chains: Polymeric protein signals. Curr. Opin. Chem. Biol. 2004, 8, 610–616. [Google Scholar] [CrossRef]
- Banh, R.S.; Iorio, C.; Marcotte, R.; Xu, Y.; Cojocari, D.; Rahman, A.A.; Pawling, J.; Zhang, W.; Sinha, A.; Rose, C.M. PTP1B controls non-mitochondrial oxygen consumption by regulating RNF213 to promote tumour survival during hypoxia. Nat. Cell Biol. 2016, 18, 803–813. [Google Scholar] [CrossRef]
- Bai, Y.; Sun, Q. Macrophage recruitment in obese adipose tissue. Obes. Rev. 2015, 16, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Song, D.D.; Chen, Y.; Li, Z.Y.; Guan, Y.F.; Zou, D.J.; Miao, C.Y. Protein tyrosine phosphatase 1B inhibits adipocyte differentiation and mediates TNFα action in obesity. Biochim. Biophys. Acta. 2013, 1831, 1368–1376. [Google Scholar] [CrossRef]
- Bansal, S.; Mahendiratta, S.; Agrawal, M.; Kumar, S.; Sharma, A.R.; Garg, N.; Joshi, R.; Sarma, P.; Prakash, A.; Chopra, K.; et al. Role of protein tyrosine phosphatase 1B inhibitor in central insulin resistance and associated cognitive deficits. Brain Res. Bull. 2021, 171, 113–125. [Google Scholar] [CrossRef]
- Lorenzo, M.; Fernández-Veledo, S.; Vila-Bedmar, R.; Garcia-Guerra, L.; De Alvaro, C.; Nieto-Vazquez, I. Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. J. Anim. Sci. 2008, 86 (Suppl. S14), E94–E104. [Google Scholar] [CrossRef]
- Ao, C.; Huo, Y.; Qi, L.; Xiong, Z.; Xue, L.; Qi, Y. Pioglitazone suppresses the lipopolysaccharide-induced production of inflammatory factors in mouse macrophages by inactivating NF-kappaB. Cell Biol. Int. 2010, 34, 723–730. [Google Scholar] [CrossRef]
- Dadke, S.S.; Li, H.C.; Kusari, A.B.; Begum, N.; Kusari, J. Elevated expression and activity of protein-tyrosine phosphatase 1B in skeletal muscle of insulin-resistant type II diabetic Goto-Kakizaki rats. Biochem. Biophys. Res. Commun. 2000, 274, 583–589. [Google Scholar] [CrossRef]
- Lam, N.T.; Covey, S.D.; Lewis, J.T.; Oosman, S.; Webber, T.; Hsu, E.C.; Cheung, A.T.; Kieffer, T.J. Leptin resistance following over-expression of protein tyrosine phosphatase 1B in liver. J. Mol. Endocrinol. 2006, 36, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Thery, F.; Martina, L.; Asselman, C.; Zhang, Y.; Vessely, M.; Repo, H.; Sedeyn, K.; Moschonas, G.D.; Bredow, C.; Teo, Q.W.; et al. Ring finger protein 213 assembles into a sensor for ISGylated proteins with antimicrobial activity. Nat. Commun. 2021, 12, 5772. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Yang, Y.C.; Hsieh, M.Y.; Yeh, Y.C.; Li, T.K. A negative feedback of the HIF-1α pathway via interferon-stimulated gene 15 and ISGylation. Clin. Cancer Res. 2013, 19, 5927–5939. [Google Scholar] [CrossRef] [Green Version]
- Brookheart, R.T.; Michel, C.I.; Schaffer, J.E. As a matter of fat. Cell Metab. 2009, 10, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Paumen, M.B.; Ishida, Y.; Muramatsu, M.; Yamamoto, M.; Honjo, T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J. Biol. Chem. 1997, 272, 3324–3329. [Google Scholar] [CrossRef] [Green Version]
- Piccolis, M.; Bond, L.M.; Kampmann, M.; Pulimeno, P.; Chitraju, C.; Jayson, C.; Vaites, L.P.; Boland, S.; Lai, Z.W.; Gabriel, K.R.; et al. Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol. Cell 2019, 74, 32–44.e8. [Google Scholar] [CrossRef] [Green Version]
- Kitai, Y.; Ariyama, H.; Kono, N.; Oikawa, D.; Iwawaki, T.; Arai, H. Membrane lipid saturation activates IRE1α without inducing clustering. Genes Cells 2013, 18, 798–809. [Google Scholar] [CrossRef]
- Sugihara, M.; Morito, D.; Ainuki, S.; Hirano, Y.; Ogino, K.; Kitamura, A.; Hirata, H.; Nagata, K. The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. J. Cell Biol. 2019, 218, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.M.; Smith, E.R. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009, 360, 1226–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mineharu, Y.; Miyamoto, S. RNF213 and GUCY1A3 in Moyamoya Disease: Key Regulators of Metabolism, Inflammation, and Vascular Stability. Front. Neurol. 2021, 12, 687088. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Son, S.M.; Mook-Jung, I.; Moon, Y.J.; Lee, J.Y.; Wang, K.C.; Kang, H.S.; Phi, J.H.; Choi, S.A.; Chong, S. Mitochondrial abnormalities related to the dysfunction of circulating endothelial colony-forming cells in moyamoya disease. J. Neurosurg. 2018, 129, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Han, C.; Jia, Y.; Wang, J.; Ge, W.; Duan, L. Proteomic Profiling of Exosomes from Hemorrhagic Moyamoya Disease and Dysfunction of Mitochondria in Endothelial Cells. Stroke 2021, 52, 3351–3361. [Google Scholar] [CrossRef]
- Geng, C.; Cui, C.; Guo, Y.; Wang, C.; Zhang, J.; Han, W.; Jin, F.; Chen, D.; Jiang, P. Metabolomic Profiling Revealed Potential Biomarkers in Patients with Moyamoya Disease. Front. Neurosci. 2020, 14, 308. [Google Scholar] [CrossRef] [Green Version]
- Dei Cas, M.; Carrozzini, T.; Pollaci, G.; Potenza, A.; Nava, S.; Canavero, I.; Tinelli, F.; Gorla, G.; Vetrano, I.G.; Acerbi, F.; et al. Plasma Lipid Profiling Contributes to Untangle the Complexity of Moyamoya Arteriopathy. Int. J. Mol. Sci. 2021, 22, 13410. [Google Scholar] [CrossRef]
- Tinelli, F.; Nava, S.; Arioli, F.; Bedini, G.; Scelzo, E.; Lisini, D.; Faragò, G.; Gioppo, A.; Ciceri, E.F.; Acerbi, F. Vascular Remodeling in Moyamoya Angiopathy: From Peripheral Blood Mononuclear Cells to Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 5763. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Houzelstein, D.; Simon-Chazottes, D.; Batista, L.; Tokuda, S.; Langa Vives, F.; Flamand, M.; Montagutelli, X.; Panthier, J.J. The ring finger protein 213 gene (Rnf213) contributes to Rift Valley fever resistance in mice. Mamm. Genome 2021, 32, 30–37. [Google Scholar] [CrossRef]
- Shraim, M.A.; Eid, R.; Radad, K.; Saeed, N. Ultrastructural pathology of human liver in Rift Valley fever. BMJ Case Rep. 2016, 2016, bcr2016216054. [Google Scholar] [CrossRef]
- Batista, L.; Jouvion, G.; Simon-Chazottes, D.; Houzelstein, D.; Burlen-Defranoux, O.; Boissière, M.; Tokuda, S.; do Valle, T.Z.; Cumano, A.; Flamand, M.; et al. Genetic dissection of Rift Valley fever pathogenesis: Rvfs2 locus on mouse chromosome 11 enables survival to early-onset hepatitis. Sci. Rep. 2020, 10, 8734. [Google Scholar] [CrossRef]
- Smith, D.R.; Steele, K.E.; Shamblin, J.; Honko, A.; Johnson, J.; Reed, C.; Kennedy, M.; Chapman, J.L.; Hensley, L.E. The pathogenesis of Rift Valley fever virus in the mouse model. Virology 2010, 407, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Reed, C.; Steele, K.E.; Honko, A.; Shamblin, J.; Hensley, L.E.; Smith, D.R. Ultrastructural study of Rift Valley fever virus in the mouse model. Virology 2012, 431, 58–70. [Google Scholar] [CrossRef] [Green Version]
- Otten, E.G.; Werner, E.; Crespillo-Casado, A.; Boyle, K.B.; Dharamdasani, V.; Pathe, C.; Santhanam, B.; Randow, F. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 2021, 594, 111–116. [Google Scholar] [CrossRef]
- Huang, J.; Brumell, J.H. Bacteria-autophagy interplay: A battle for survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef]
- Perrin, A.J.; Jiang, X.; Birmingham, C.L.; So, N.S.; Brumell, J.H. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr. Biol. 2004, 14, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001, 20, 362–371. [Google Scholar] [CrossRef]
- Zhao, C.; Beaudenon, S.L.; Kelley, M.L.; Waddell, M.B.; Yuan, W.; Schulman, B.A.; Huibregtse, J.M.; Krug, R.M. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc. Natl. Acad. Sci. USA 2004, 101, 7578–7582. [Google Scholar] [CrossRef] [Green Version]
- Dastur, A.; Beaudenon, S.; Kelley, M.; Krug, R.M.; Huibregtse, J.M. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J. Biol. Chem. 2006, 281, 4334–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, F.; Zou, W.; Zhang, D.E. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev. 2007, 21, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Impens, F.; Ribet, D.; Quereda, J.J.; Nam Tham, T.; Nahori, M.A.; Bierne, H.; Dussurget, O.; Pizarro-Cerdá, J.; Knobeloch, K.P.; et al. ISG15 counteracts Listeria monocytogenes infection. eLife 2015, 4, e06848. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.; Malakhov, M.; Hetherington, C.; Zhang, D.E. Lipopolysaccharide activates the expression of ISG15-specific protease UBP43 via interferon regulatory factor 3. J. Biol. Chem. 2002, 277, 14703–14711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzigher, L.; Pavlovic, J.; Haller, O.; Staeheli, P. Mx genes show weaker primary response to virus than other interferon-regulated genes. Virology 1992, 186, 154–160. [Google Scholar] [CrossRef]
- Kimmey, J.M.; Campbell, J.A.; Weiss, L.A.; Monte, K.J.; Lenschow, D.J.; Stallings, C.L. The impact of ISGylation during Mycobacterium tuberculosis infection in mice. Microbes Infect. 2017, 19, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, A.; van der Veen, A.G.; Bunyan, M.; Borg, A.; Frith, D.; Howell, S.; Kjaer, S.; Beling, A.; Snijders, A.P.; Knobeloch, K.-P.; et al. Cysteine-Reactive Free ISG15 Generates IL-1β-Producing CD8α+ Dendritic Cells at the Site of Infection. J. Immunol. 2018, 201, 604–614. [Google Scholar] [CrossRef] [Green Version]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Cecchi, A.C.; Guo, D.; Ren, Z.; Flynn, K.; Santos-Cortez, R.L.; Leal, S.M.; Wang, G.T.; Regalado, E.S.; Steinberg, G.K.; Shendure, J.; et al. RNF213 rare variants in an ethnically diverse population with Moyamoya disease. Stroke 2014, 45, 3200–3207. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, A.; Kobayashi, H.; Liu, W.; Fujii, Y.; Senevirathna, S.T.; Nanayakkara, S.; Okuda, H.; Hitomi, T.; Harada, K.H.; Takenaka, K.; et al. R4810K, a polymorphism of RNF213, the susceptibility gene for moyamoya disease, is associated with blood pressure. Environ. Health Prev. Med. 2013, 18, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Liang, J.; Wen, J.; Luo, M.; Li, J.; Sun, X.; Xu, X.; Li, J.; Wang, D.; Wang, J.; et al. Mutations of RNF213 are responsible for sporadic cerebral cavernous malformation and lead to a mulberry-like cluster in zebrafish. J. Cereb. Blood Flow. Metab. 2021, 41, 1251–1263. [Google Scholar] [CrossRef]
- Kiando, S.R.; Barlassina, C.; Cusi, D.; Galan, P.; Lathrop, M.; Plouin, P.F.; Jeunemaitre, X.; Bouatia-Naji, N. Exome sequencing in seven families and gene-based association studies indicate genetic heterogeneity and suggest possible candidates for fibromuscular dysplasia. J. Hypertens. 2015, 33, 1802–1810. [Google Scholar] [CrossRef]
- Miyawaki, S.; Imai, H.; Shimizu, M.; Yagi, S.; Ono, H.; Mukasa, A.; Nakatomi, H.; Shimizu, T.; Saito, N. Genetic variant RNF213 c.14576G>A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke 2013, 44, 2894–2897. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Ambalavanan, A.; Rochefort, D.; Xie, P.; Bourassa, C.V.; Hince, P.; Dionne-Laporte, A.; Spiegelman, D.; Gan-Or, Z.; Mirarchi, C.; et al. RNF213 Is Associated with Intracranial Aneurysms in the French-Canadian Population. Am. J. Hum. Genet. 2016, 99, 1072–1085. [Google Scholar] [CrossRef] [Green Version]
- Sigdel, T.K.; Shoemaker, L.D.; Chen, R.; Li, L.; Butte, A.J.; Sarwal, M.M.; Steinberg, G.K. Immune response profiling identifies autoantibodies specific to Moyamoya patients. Orphanet. J. Rare Dis. 2013, 8, 45. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollaci, G.; Gorla, G.; Potenza, A.; Carrozzini, T.; Canavero, I.; Bersano, A.; Gatti, L. Novel Multifaceted Roles for RNF213 Protein. Int. J. Mol. Sci. 2022, 23, 4492. https://doi.org/10.3390/ijms23094492
Pollaci G, Gorla G, Potenza A, Carrozzini T, Canavero I, Bersano A, Gatti L. Novel Multifaceted Roles for RNF213 Protein. International Journal of Molecular Sciences. 2022; 23(9):4492. https://doi.org/10.3390/ijms23094492
Chicago/Turabian StylePollaci, Giuliana, Gemma Gorla, Antonella Potenza, Tatiana Carrozzini, Isabella Canavero, Anna Bersano, and Laura Gatti. 2022. "Novel Multifaceted Roles for RNF213 Protein" International Journal of Molecular Sciences 23, no. 9: 4492. https://doi.org/10.3390/ijms23094492
APA StylePollaci, G., Gorla, G., Potenza, A., Carrozzini, T., Canavero, I., Bersano, A., & Gatti, L. (2022). Novel Multifaceted Roles for RNF213 Protein. International Journal of Molecular Sciences, 23(9), 4492. https://doi.org/10.3390/ijms23094492