
The Discovery of Potent SHP2 Inhibitors with Anti-Proliferative Activity in Breast Cancer Cell Lines

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
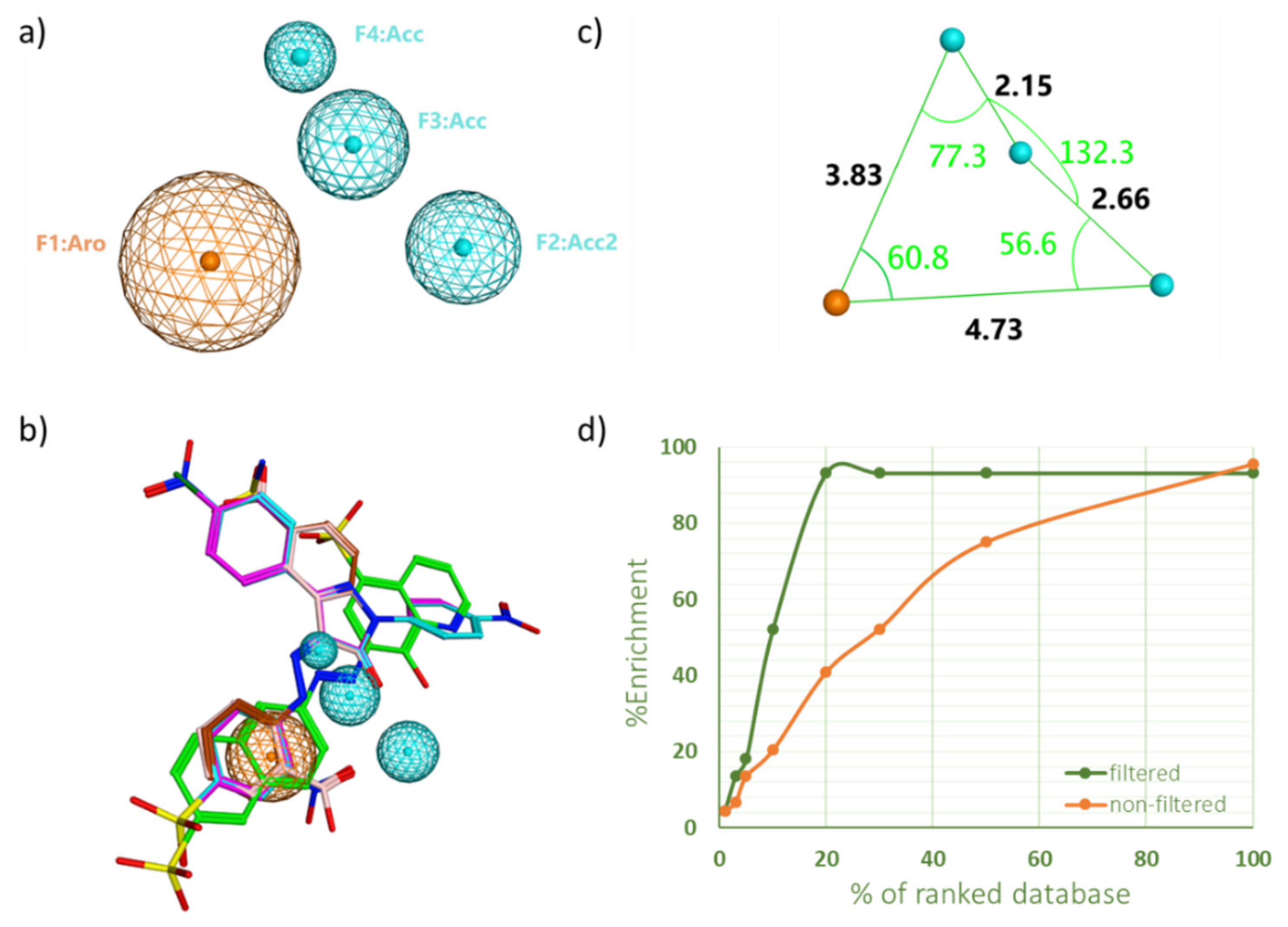
2.1. Pharmacophore Model Generation and Validation
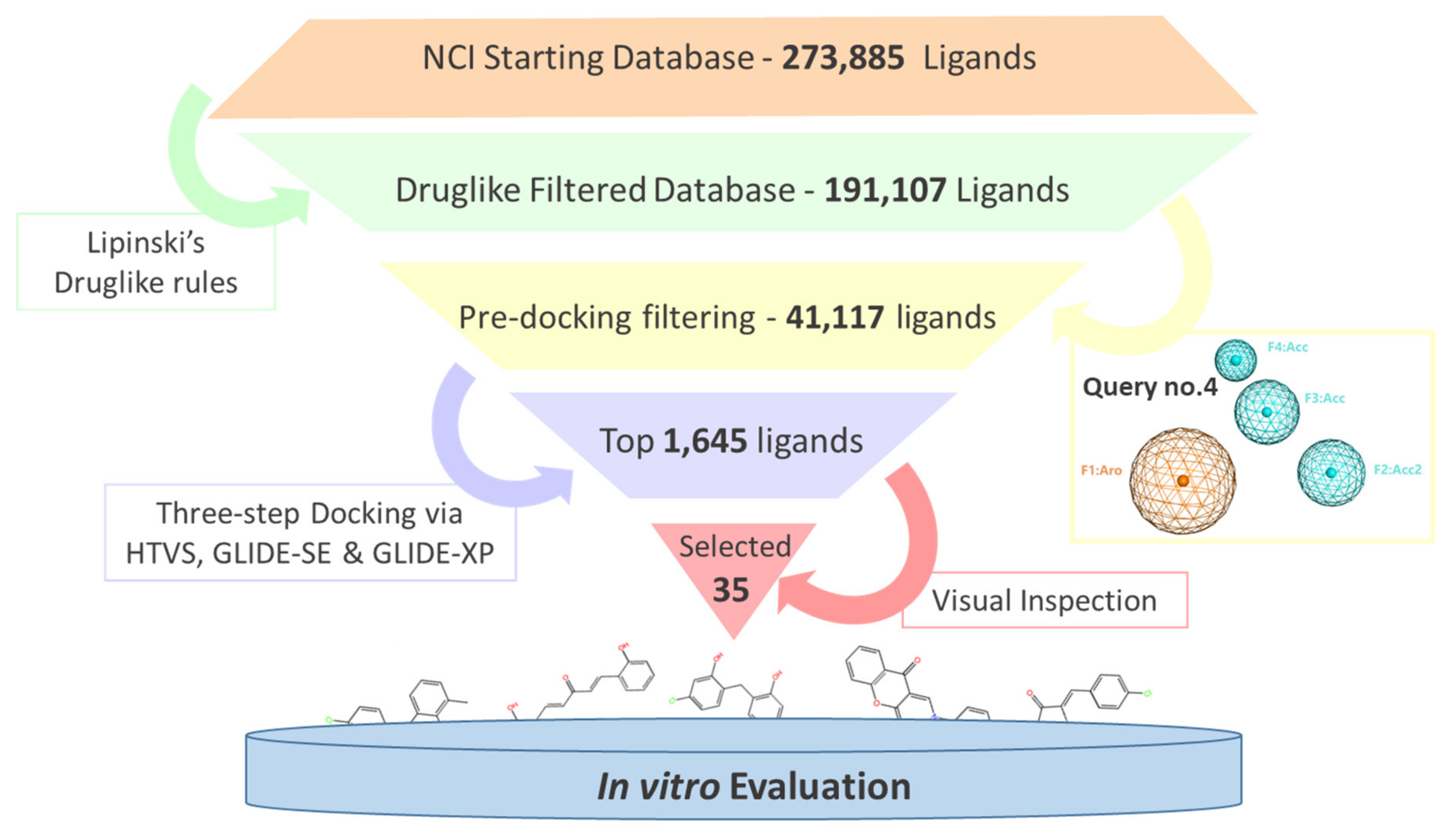
2.2. Virtual Screening and Compounds Selection
2.3. Inhibition of SHP2 Enzyme Activity by the Compounds NSC 13030, 24198, 57774, and 137420
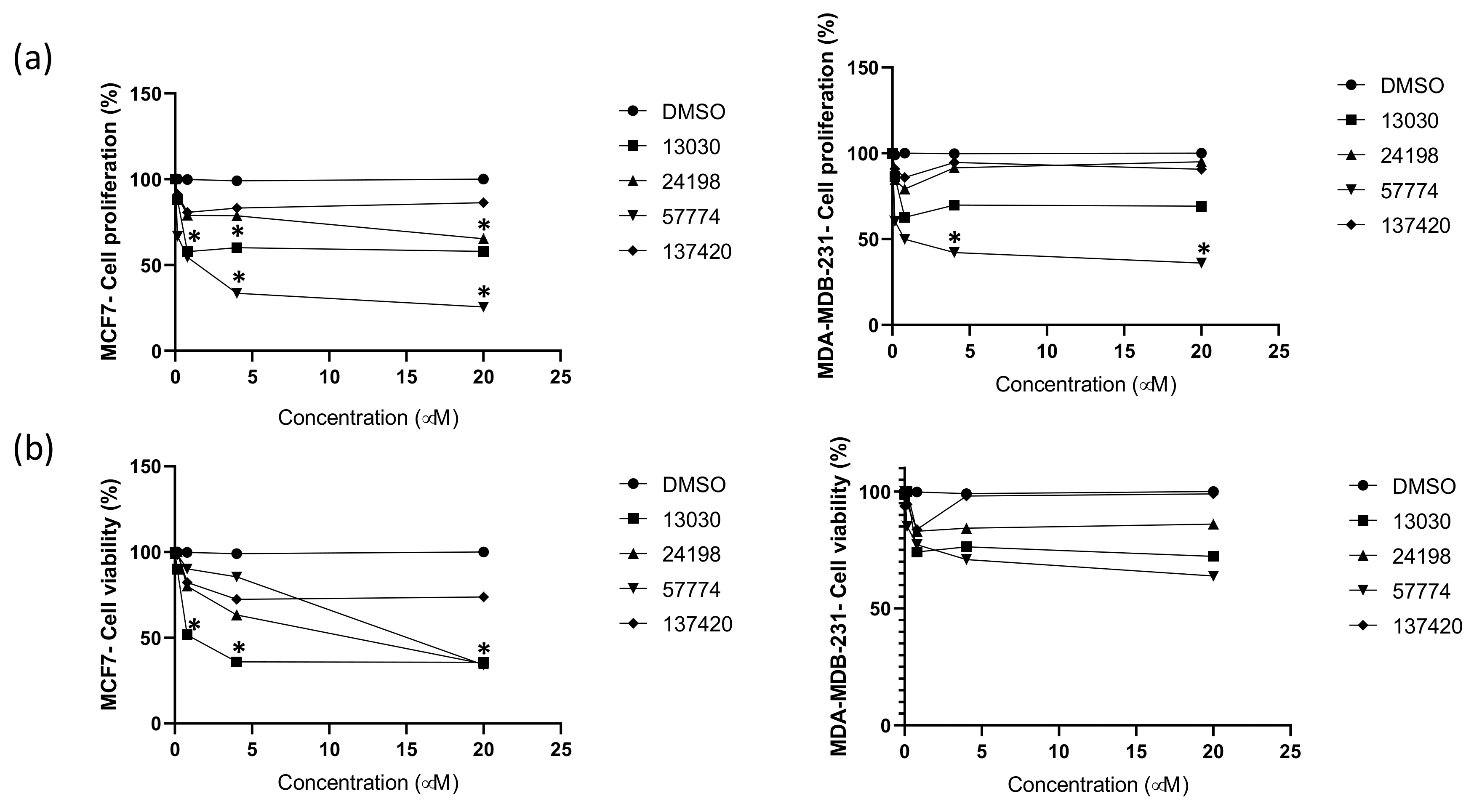
2.4. Compounds Inhibit Breast Cancer Cells’ Proliferation and Viability
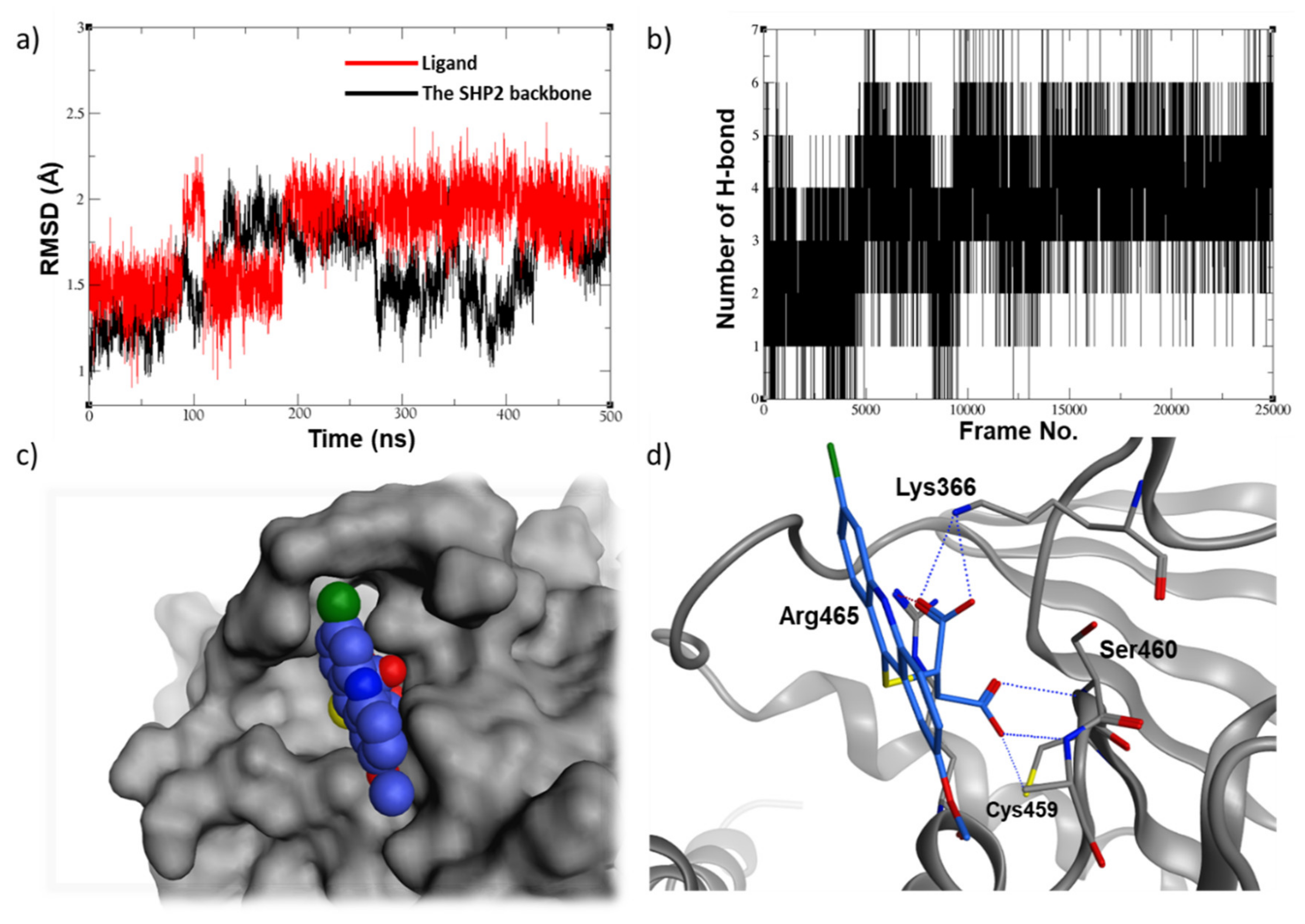
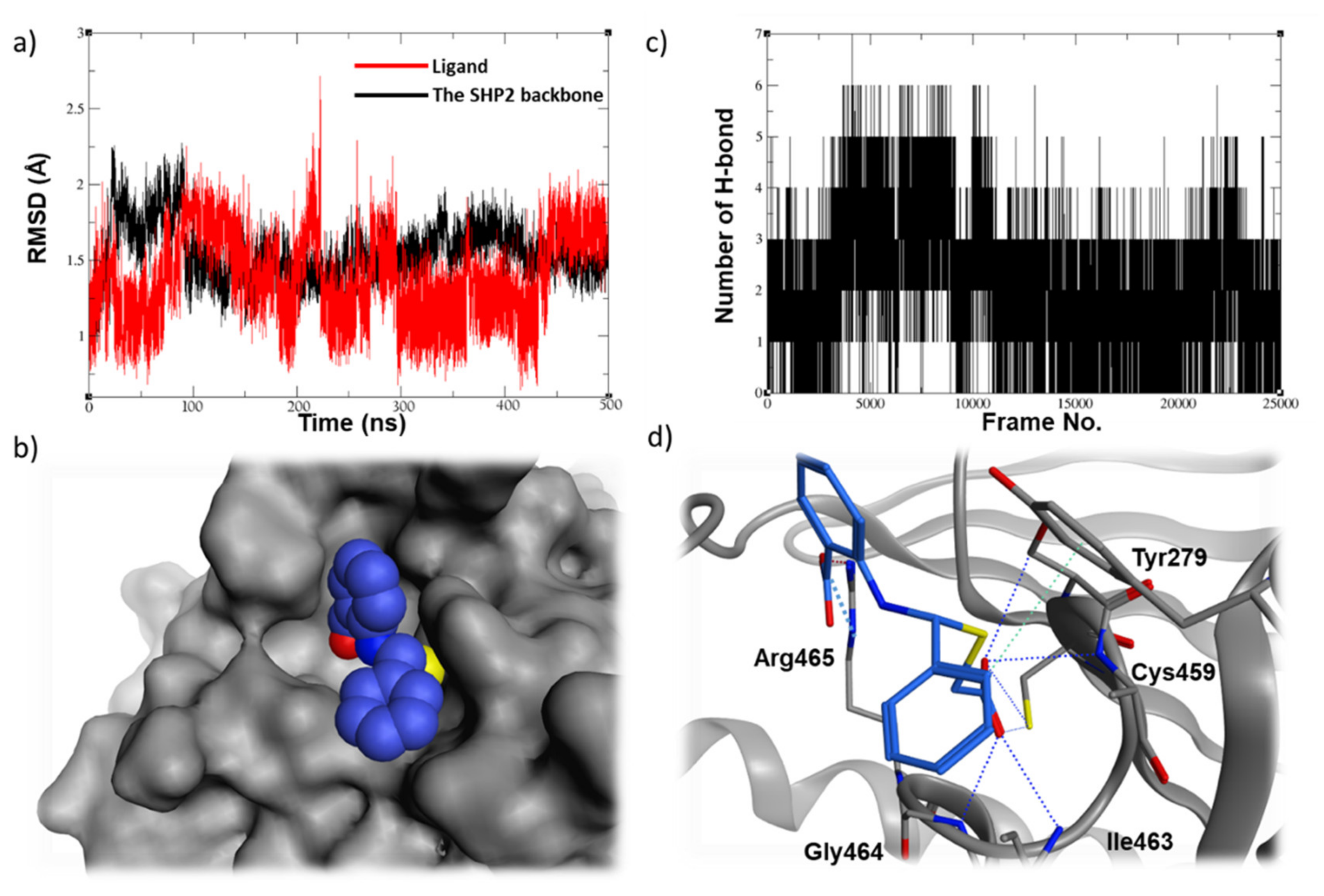
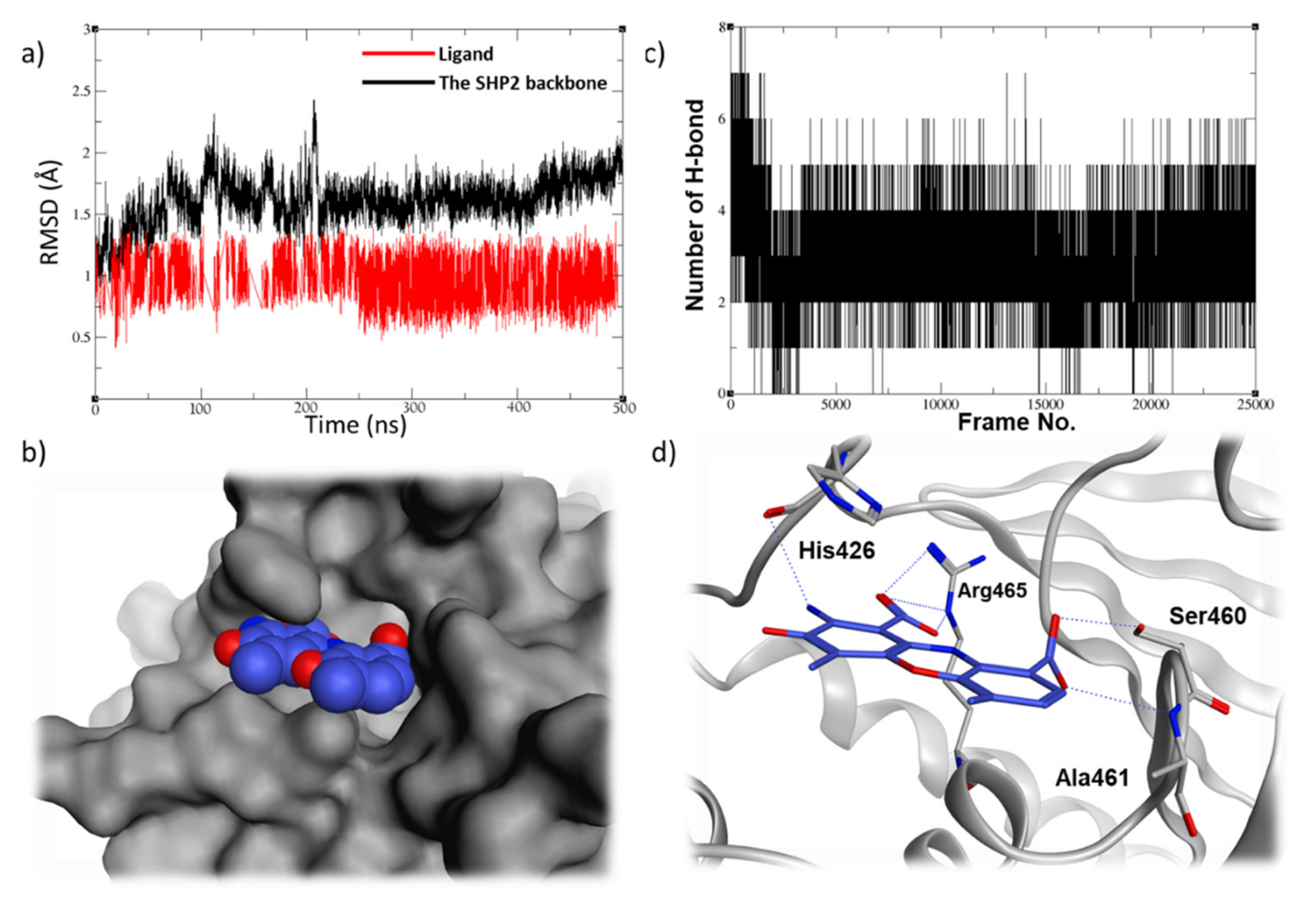
2.5. Molecular Dynamic Studies for Compounds Stability and Fitting inside the SHP2 Active Site
2.6. Assessment of Pharmacokinetic and Druglike Characteristics
3. Materials and Methods
3.1. Literature Review
3.2. Pharmacophore Generation and Validation
3.3. Validation of the Virtual Screening Protocol
3.4. Virtual Screening
3.5. Molecular Dynamic (MD) Simulations
3.6. Clustering Analysis
3.7. Cell Proliferative Assay
3.8. Cell Viability Assay
3.9. Enzymatic SHP2 Inhibition Assay
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 20 November 2021).
- Ginsburg, O.; Bray, F.; Coleman, M.P.; Vanderpuye, V.; Eniu, A.; Kotha, R.; Sarker, M.; Huong, T.T.; Allemani, C.; Dvaladze, A.; et al. The Global Burden of Women’s Cancers: An Unmet Grand Challenge in Global Health Europe PMC Funders Group. Lancet 2017, 389, 847–860. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Tyrosine Phosphorylation: Thirty Years and Counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Protein Tyrosine Phosphatases: Prospects for Therapeutics. Curr. Opin. Chem. Biol. 2001, 5, 416–423. [Google Scholar] [CrossRef]
- Julien, S.G.; Dubé, N.; Hardy, S.; Tremblay, M.L. Inside the Human Cancer Tyrosine Phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Van Etten, R.A. Tyrosine Kinases as Targets for Cancer Therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Ventura, J.-J.; Nebreda, A.R. Protein Kinases and Phosphatases as Therapeutic Targets in Cancer. Clin. Transl. Oncol. 2006, 8, 153–160. [Google Scholar] [CrossRef]
- Sivaganesh, V.; Sivaganesh, V.; Scanlon, C.; Iskander, A.; Maher, S.; Lê, T.; Peethambaran, B. Protein Tyrosine Phosphatases: Mechanisms in Cancer. Int. J. Mol. Sci. 2021, 22, 12865. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in Cancer. J. Cell. Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- Liu, Q.; Qu, J.; Zhao, M.; Xu, Q.; Sun, Y. Targeting SHP2 as a Promising Strategy for Cancer Immunotherapy. Pharmacol. Res. 2020, 152, 104595. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS Nucleotide Cycling Underlies the SHP2 Phosphatase Dependence of Mutant BRAF-, NF1- and RAS-Driven Cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating Mutations of the Noonan Syndrome-Associated SHP2/PTPN11 Gene in Human Solid Tumors and Adult Acute Myelogenous Leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.; Miyamoto, M.; Takahashi, A.; Yomogita, Y.; Higashi, H.; Kondo, S.; Hatakeyama, M. Isolation of a Distinct Class of Gain-of-Function SHP-2 Mutants with Oncogenic RAS-like Transforming Activity from Solid Tumors. Oncogene 2008, 27, 3508–3515. [Google Scholar] [CrossRef]
- Dong, L.; Han, D.; Meng, X.; Xu, M.; Zheng, C.; Xia, Q. Activating Mutation of SHP2 Establishes a Tumorigenic Phonotype Through Cell-Autonomous and Non-Cell-Autonomous Mechanisms. Front. Cell Dev. Biol. 2021, 9, 630712. [Google Scholar] [CrossRef]
- Song, M.; Park, J.E.; Park, S.G.; Lee, D.H.; Choi, H.K.; Park, B.C.; Ryu, S.E.; Kim, J.H.; Cho, S. NSC-87877, Inhibitor of SHP-1/2 PTPs, Inhibits Dual-Specificity Phosphatase 26 (DUSP26). Biochem. Biophys. Res. Commun. 2009, 381, 491–495. [Google Scholar] [CrossRef]
- Chen, Y.N.P.; Lamarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.T.; Chen, Z.; Cooke, V.G.; et al. Allosteric Inhibition of SHP2 Phosphatase Inhibits Cancers Driven by Receptor Tyrosine Kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Peach, M.L.; Nicklaus, M.C. Combining Docking with Pharmacophore Filtering for Improved Virtual Screening. J. Cheminform. 2009, 1, 6. [Google Scholar] [CrossRef]
- Gupta, C.L.; Babu Khan, M.; Ampasala, D.R.; Akhtar, S.; Dwivedi, U.N.; Bajpai, P. Pharmacophore-Based Virtual Screening Approach for Identification of Potent Natural Modulatory Compounds of Human Toll-like Receptor 7. J. Biomol. Struct. Dyn. 2019, 37, 4721–4736. [Google Scholar] [CrossRef]
- Wu, J.; Li, W.; Zheng, Z.; Lu, X.; Zhang, H.; Ma, Y.; Wang, R. Design, Synthesis, Biological Evaluation, Common Feature Pharmacophore Model and Molecular Dynamics Simulation Studies of Ethyl 4-(Phenoxymethyl)-2-Phenylthiazole-5-Carboxylate as Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (SHP2) Inhi. J. Biomol. Struct. Dyn. 2021, 39, 1174–1188. [Google Scholar] [CrossRef]
- Craggs, G.; Kellie, S. A Functional Nuclear Localization Sequence in the C-Terminal Domain of SHP-1. J. Biol. Chem. 2001, 276, 23719–23725. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Libring, S.; Ruddraraju, K.V.; Miao, J.; Solorio, L.; Zhang, Z.Y.; Wendt, M.K. SHP2 Is a Multifunctional Therapeutic Target in Drug Resistant Metastatic Breast Cancer. Oncogene 2020, 39, 7166–7180. [Google Scholar] [CrossRef] [PubMed]
- Grubczak, K.; Kretowska-grunwald, A.; Groth, D.; Poplawska, I.; Kruszewska, J.; Wojtukiewicz, M.Z.; Moniuszko, M. Differential Response of MDA-MB-231 and MCF-7 Breast Cancer Cells to In Vitro Inhibition with CTLA-4 and PD-1 through Cancer-Immune Cells Modified Interactions. Cells 2021, 10, 2044. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, N.; Karthikeyan, C.; Bhadada, S.V.; Verma, A.K.; Sahi, C.; Moorthy, N.S.H.N.; Trivedi, P. Design, Synthesis and Biological Evaluation of Some Tetrazole Acetamide Derivatives as Novel Non-Carboxylic PTP1B Inhibitors. Bioorg. Chem. 2019, 92, 103221. [Google Scholar] [CrossRef] [PubMed]
- Kousaxidis, A.; Petrou, A.; Lavrentaki, V.; Fesatidou, M.; Nicolaou, I.; Geronikaki, A. Aldose Reductase and Protein Tyrosine Phosphatase 1B Inhibitors as a Promising Therapeutic Approach for Diabetes Mellitus. Eur. J. Med. Chem. 2020, 207, 112742. [Google Scholar] [CrossRef]
- Walunj, D.; Egarmina, K.; Tuchinsky, H.; Shpilberg, O.; Hershkovitz-Rokah, O.; Grynszpan, F.; Gellerman, G. Expedient Synthesis and Anticancer Evaluation of Dual-Action 9-Anilinoacridine Methyl Triazene Chimeras. Chem. Biol. Drug Des. 2021, 97, 237–252. [Google Scholar] [CrossRef]
- Kalirajan, R.; Gaurav, K.; Pandiselvi, A.; Gowramma, B.; Sankar, S. Novel Thiazine Substituted 9-Anilinoacridines: Synthesis, Antitumour Activity and Structure Activity Relationships. Anticancer. Agents Med. Chem. 2019, 19, 1350–1358. [Google Scholar] [CrossRef]
- Khunnawutmanotham, N.; Jumpathong, W.; Eurtivong, C.; Chimnoi, N.; Techasakul, S. Synthesis, Cytotoxicity Evaluation, and Molecular Modeling Studies of 2, N10-Substituted Acridones as DNA-Intercalating Agents. J. Chem. Res. 2020, 44, 410–425. [Google Scholar] [CrossRef]
- Bereznyak, E.G.; Semenov, M.A.; Bol’bukh, T.V.; Dukhopel’nikov, E.V.; Shestopalova, A.V.; Maleev, V.Y. A Study of the Effect of Water on the Interaction of DNA with Actinocin Derivatives Having Different Lengths of Aminoalkyl Chains by the Methods of IR Spectroscopy and Computer Simulation. Biofizika 2002, 47, 1014. [Google Scholar]
- Maleev, V.; Semenov, M.; Kruglova, E.; Bolbukh, T.; Gasan, A.; Bereznyak, E.; Shestopalova, A. Spectroscopic and Calorimetric Study of DNA Interaction with a New Series of Actinocin Derivatives. J. Mol. Struct. 2003, 645, 145–158. [Google Scholar] [CrossRef]
- Miroshnychenko, K.V.; Shestopalova, A.V. Flexible Docking of DNA Fragments and Actinocin Derivatives. Mol. Simul. 2005, 31, 567–574. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- McGovern, S.; Caselli, E.; Grigorieff, N.; Shoichet, K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE). 2019. Available online: https://www.chemcomp.com/products.htm (accessed on 20 March 2022).
- Rapp, C.S.; Schonbrun, C.; Jacobson, M.P.; Kalyanaraman, C.; Huang, N. Automated Site Preparation in Physics-Based Rescoring of Receptor Ligand Complexes. Proteins 2009, 77, 52–61. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical Method Validation: New FDA Guidance vs. EMA Guideline. Better or Worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Yu, Z.H.; Zhang, R.Y.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Shim, J.S.; Zeng, L.F.; He, Y.; Chen, L.; et al. Exploring the Existing Drug Space for Novel PTyr Mimetic and SHP2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, A.; Nandi, S.P. Identification of Potent Inhibitors of Covid-19 Main Protease Enzyme by Molecular Docking Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Schrödinger. 2021. Available online: https://www.schrodinger.com/products/ligprep (accessed on 20 March 2022).
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Lee, T.S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; Dwight McGee, T.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Guimarães, C.R.W.; Cardozo, M. MM-GB/SA Rescoring of Docking Poses in Structure-Based Lead Optimization. J. Chem. Inf. Model. 2008, 48, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
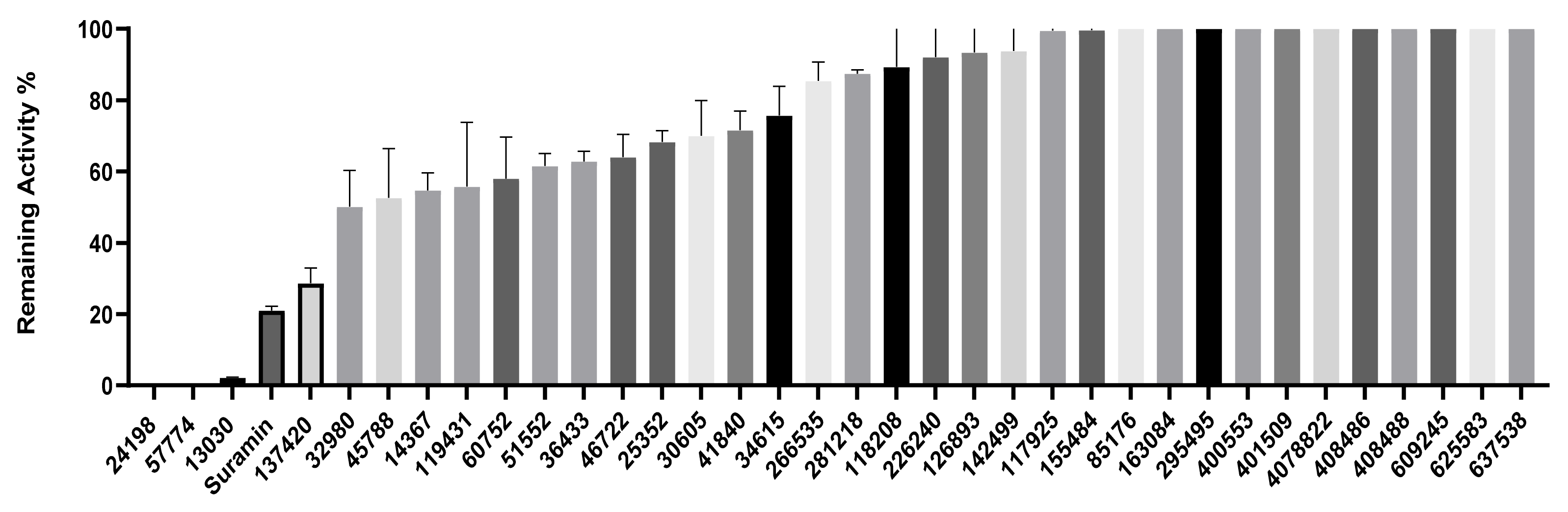{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PH4 No. | Features | Selectivity Se (%) | Specificity Sp (%) |
|---|---|---|---|
| 4 | Aro Acc2 Acc Acc | 100 | 90 |
| 7 | Aro Aro Acc2 Acc | 93.2 | 93.3 |
| 22 | Aro Hyd Acc2 Acc | 93.18 | 66.67 |
| 30 | Hyd Hyd Acc Acc | 84.09 | 80 |
| 39 | Hyd Acc2 Acc Acc | 97.72 | 56.66 |
| 42 | Aro Hyd Acc2 Acc | 95.45 | 60 |
| Top Ranked Portions of the Docked Ligand Library | % Enrichment (% EF) | |
|---|---|---|
| SVS | FVS | |
| 1% | 4.5 | 4.5 |
| 3% | 6.8 | 13.6 |
| 5% | 13.6 | 18.2 |
| 10% | 20.5 | 52.3 |
| 20% | 40.9 | 95.5 |
| Compound ID | Chemical Structures | SHP2 | SHP1 |
|---|---|---|---|
| Obtained IC50 (μM) | |||
| Suramin |  | 4.5 | 15.4 |
| 13030 |  | 3.2 | 85.4 |
| 24198 |  | 1.9 | 14.3 |
| 57774 |  | 0.8 | 164.4 |
| 137420 |  | 33 | NT |
| Compound ID | |||||
|---|---|---|---|---|---|
| 13030 | 24198 | 57774 | 137420 | ||
| Pharmacokinetics | GI absorption | High | High | Low | High |
| BBB permeant | No | No | No | No | |
| CYP1A2 inhibitor | Yes | No | No | No | |
| CYP2C19 inhibitor | Yes | No | No | No | |
| CYP2C9 inhibitor | Yes | Yes | No | No | |
| CYP2D6 inhibitor | No | No | No | No | |
| CYP3A4 inhibitor | No | No | No | No | |
| Bioavailability score | 0.56 | 0.56 | 0.56 | 0.56 | |
| Druglikeness | Lipinski’s rules | Yes | Yes | Yes | Yes |
| Veber’s rules | Yes | Yes | No; TPSA >140 | Yes | |
| Ghose’s rules | Yes | Yes | Yes | Yes | |
| Egan’s rules | Yes | Yes | No; TPSA > 131.6 | Yes | |
| Muegge’s rules | Yes | Yes | Yes | Yes | |
| Leadlikeness | PAINS | No alert | No alert | No alert | No alert |
| Brenk’s rules | 1 alert; (polycyclic_aromatichydrocarbon_2 | 1 alert; (het_C_het_not_in_ring) | 2 alerts; (aniline, polycyclic_aromatic_hydrocarbon_2) | No alert | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghemrawi, R.; Khair, M.; Hasan, S.; Aldulaymi, R.; AlNeyadi, S.S.; Atatreh, N.; Ghattas, M.A. The Discovery of Potent SHP2 Inhibitors with Anti-Proliferative Activity in Breast Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 4468. https://doi.org/10.3390/ijms23084468
Ghemrawi R, Khair M, Hasan S, Aldulaymi R, AlNeyadi SS, Atatreh N, Ghattas MA. The Discovery of Potent SHP2 Inhibitors with Anti-Proliferative Activity in Breast Cancer Cell Lines. International Journal of Molecular Sciences. 2022; 23(8):4468. https://doi.org/10.3390/ijms23084468
Chicago/Turabian StyleGhemrawi, Rose, Mostafa Khair, Shaima Hasan, Raghad Aldulaymi, Shaikha S. AlNeyadi, Noor Atatreh, and Mohammad A. Ghattas. 2022. "The Discovery of Potent SHP2 Inhibitors with Anti-Proliferative Activity in Breast Cancer Cell Lines" International Journal of Molecular Sciences 23, no. 8: 4468. https://doi.org/10.3390/ijms23084468
APA StyleGhemrawi, R., Khair, M., Hasan, S., Aldulaymi, R., AlNeyadi, S. S., Atatreh, N., & Ghattas, M. A. (2022). The Discovery of Potent SHP2 Inhibitors with Anti-Proliferative Activity in Breast Cancer Cell Lines. International Journal of Molecular Sciences, 23(8), 4468. https://doi.org/10.3390/ijms23084468